Abstract
Background. Periodontal diseases have been associated with several systemic conditions, including certain types of cancer. Chronic inflammation, a characteristic feature of periodontal disease, has been hypothesized to contribute to carcinogenesis and tumor progression. However, the specific relationship between Aggregatibacter actinomycetemcomitans and cancer development remains an area of ongoing research.
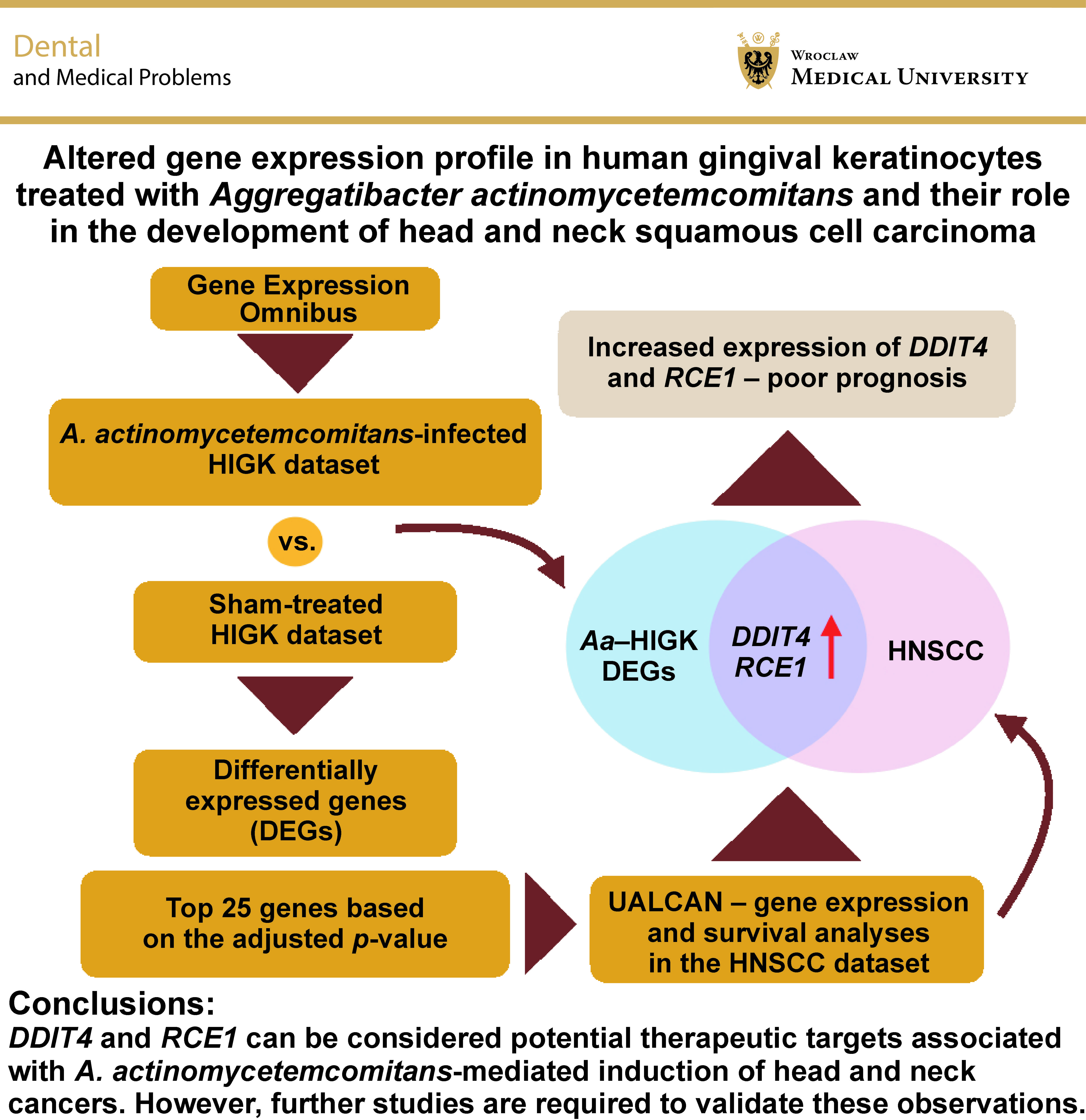
Objectives. The aim of the study was to investigate the potential association between A. actinomycetemcomitans, a well-established periodontal pathogen, and the development of head and neck squamous cell carcinoma (HNSCC). Additionally, the objective was to examine differentially expressed genes (DEGs) in A. actinomycetemcomitans-infected human immortalized gingival keratinocytes (HIGKs) and compare their expression in the HNSCC dataset using computational tools.
Material and methods. The Gene Expression Omnibus (GEO) dataset GSE9723 was used to retrieve the global gene expression profile of human gingival keratinocytes exposed to A. actinomycetemcomitans. A computational approach was employed using the PANTHER (Protein ANalysis THrough Evolutionary Relationships) Classification System for gene ontology analysis, STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) for network analysis and UALCAN (University of ALabama at Birmingham CANcer data analysis) for gene expression and survival analyses.
Results. The GEO2R analysis identified numerous DEGs, of which the top 25 were selected for further evaluation using the HNSCC dataset from The Cancer Genome Atlas (TCGA). Among these genes, DDIT4 and RCE1 demonstrated expression patterns consistent with those observed in A. actinomycetemcomitans-infected HIGKs. Further analysis revealed that patients presenting with elevated DDIT4 and RCE1 expression levels demonstrated a marked reduction in overall survival.
Conclusions. The findings suggest that DDIT4 and RCE1 can be considered potential therapeutic targets associated with A. actinomycetemcomitans-mediated induction of head and neck cancers. However, further studies are required to validate these observations.
Keywords: gene expression, periodontitis, prognosis, carcinoma, health
Introduction
Head and neck squamous cell carcinoma (HNSCC) is among the most common types of cancer worldwide, the majority of which are squamous cell carcinomas. It has an annual incidence exceeding 550,000 cases and is responsible for approx. 300,000 deaths each year.1 In India, oral squamous cell carcinoma (OSCC), a predominant subtype of HNSCC, accounts for 30% of all cancer cases. Patients diagnosed with HNSCC at an early clinical stage exhibit a survival rate of 70–90%; however, 50% of patients diagnosed at advanced stages pass away within 2 years of the initial diagnosis. One of the major clinical challenges associated with HNSCC is the lack of early screening and diagnostic procedures.2 The oral microbiome harbors diverse forms of microorganisms that adapt to different physiological and pathological conditions.3 Dysbiosis of the oral microbiota may lead to mild forms of illness, manifesting as cavities, or more serious forms, contributing to changes in normal cells. Head and neck squamous cell carcinoma has been associated with several microorganisms capable of altering host gene expression, including Haemophilus influenzae, Prevotella copri and Aggregatibacter actinomycetemcomitans. They are commensals of the oral microbiota. However, their balance may be disrupted due to habitual practices, systemic diseases and poor oral hygiene, among others.4 Aggregatibacter actinomycetemcomitans is a Gram-negative, facultatively anaerobic, capnophilic bacillus recognized as a major cause of periodontal disease in children and adolescents. It has also been associated with the development of head and neck cancer.5 Individuals with periodontitis demonstrate an increased risk of extraoral A. actinomycetemcomitans infections. Nevertheless, colonies of A. actinomycetemcomitans have also been observed in patients with good oral health and in edentulous individuals due to the organism’s ability to adhere to epithelial cells and tooth surfaces.6 Furthermore, Aggregatibacter actinomycetemcomitans has been reported to enhance bacterial internalization via phagocytosis while simultaneously limiting excessive inflammatory responses by downregulating interleukin (IL)-1 expression and reactive oxygen species (ROS) generation in macrophages.7
Teshima et al. investigated several periodontal pathogens, including Aggregatibacter actinomycetemcomitans Y4, a microaerophilic organism isolated primarily from subgingival dental plaque.8, 9 It was identified for its ability to induce double-strand breaks (DSBs) in the DNA. Notably, the accumulation of DSBs persisted even after the inhibition of apoptosis, revealing an independent mechanism. These findings suggest a possible association between certain periodontal pathogens, genome instability and an elevated risk of carcinogenesis, thereby highlighting the intricate interplay between periodontal health and oral cancer susceptibility.8, 9 Although only limited evidence is currently available regarding the role of bacterial pathogens in carcinogenesis, researchers have ventured into the exploration of bacteria-mediated induction of tumorigenesis. Computational approaches have been used for several decades to reveal the role of gene families,10 gene networks11 and epigenetic components12 in the development of cancer. The present study aimed to examine the possible association between A. actinomycetemcomitans and the initiation of head and neck tumors. The study is supported by data generated with the use of computational approaches.
Material and methods
Sample dataset
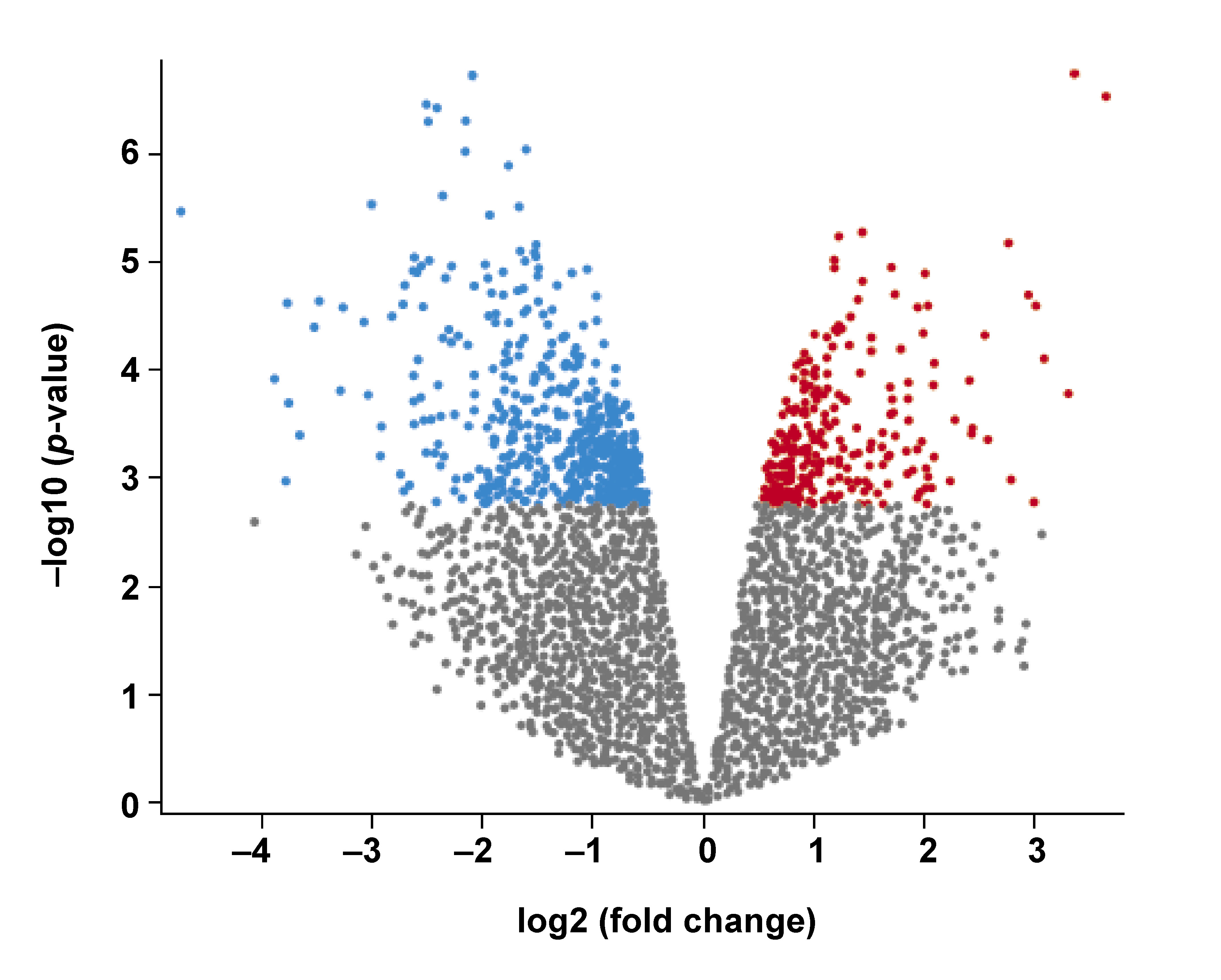
The Gene Expression Omnibus (GEO) dataset GSE9723 was used in the present study. The dataset comprised 4 samples with A. actinomycetemcomitans-infected human immortalized gingival keratinocytes (HIGKs) (GSM245733, GSM245734, GSM245735, GSM245736) and 4 Sham-infected HIGK samples (GSM245729, GSM245730, GSM245731, GSM245732).13 The Sham-infected HIGKs served as controls. The HIGKs infected with A. actinomycetemcomitans were considered the test group (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE9723). An adjusted p-value <0.05 was deemed statistically significant. As the analysis yielded an exhaustive list of differentially expressed genes (DEGs), only the top 25 DEGs were selected for further analysis (Table 1, Figure 1).
Gene expression and survival analysis
The expression levels of the top 25 DEGs retrieved from the previous analysis were investigated using the UALCAN database (http://ualcan.path.uab.edu). The analysis included 520 primary tumor HNSCC samples and 44 normal tissue samples. Gene expression levels were measured in transcripts per million (TPM), a standardized unit for RNA sequencing (RNA-seq) data normalization. Significant differences between the groups were determined using box-and-whisker plots generated from the TPM values. The Kaplan–Meier analysis was used to demonstrate overall survival of patients with HNSCC. The high-expression and low/medium-expression groups were compared to assess the effect of changes in gene expression on the overall survival of patients.14
Gene ontology analysis
The gene ontology analysis was performed using the PANTHER (Protein ANalysis THrough Evolutionary Relationships) Classification System, v. 16.0 (https://pantherdb.org). The analysis provided information regarding molecular pathways and functions, biological processes and subcellular localization of the identified genes. A user-defined query of the top 25 genes was submitted as a batch for functional classification of genes. Additionally, classification based on pathways was conducted to identify potential pathways associated with these genes.15, 16
Protein–protein interaction analysis
The protein–protein interactions between the top 25 DEGs were analyzed using the STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) tool, v. 10.0. STRING is a bioinformatics resource that provides information on protein–protein interactions, including direct physical interactions and functional associations. The nodes represent proteins interacting in a specific network, whereas edges indicate protein–protein interactions, including physical, enzymatic and genetic interactions.17
Statistical analysis
Gene expression data from multiple datasets was analyzed using the GEO2R tool. GEO2R uses the DESeq2 and Limma packages from the R programming environment to compare groups of samples within a GEO series and identify DEGs. The results are presented in the form of a table and graphic plots. The UALCAN portal was used to analyze gene expression profiles by comparing expression levels between different groups via a Perl script accompanied by a Comprehensive Perl Archive Network (CPAN) module. Survival plots were generated with the use of the survival and survminer packages in R. Differences between the groups were assessed using the log-rank test. The survival package was exclusively used for survival analysis, including the generation of survival curves, hypothesis testing and survival modeling. The use of the survminer package improved the visualization of Kaplan–Meier plots and forest plots, allowing for clearer representation and interpretation of complex survival data.14
Results
GEO analysis
Affymetrix-based microarray analysis revealed numerous DEGs. These DEGs constituted a group of genes exclusively activated or repressed upon exposure to A. actinomycetemcomitans. The volcano plot demonstrated numerous upregulated (red) and downregulated (blue) DEGs in the HIGKs infected with A. actinomycetemcomitans (Figure 1). Among the identified DEGs, the top 25 genes were curated based on adjusted p-values (Table 1). This subset included 19 upregulated genes and 6 downregulated genes, which were subsequently probed in the HNSCC dataset derived from The Cancer Genome Atlas (TCGA).
Gene expression and survival analysis
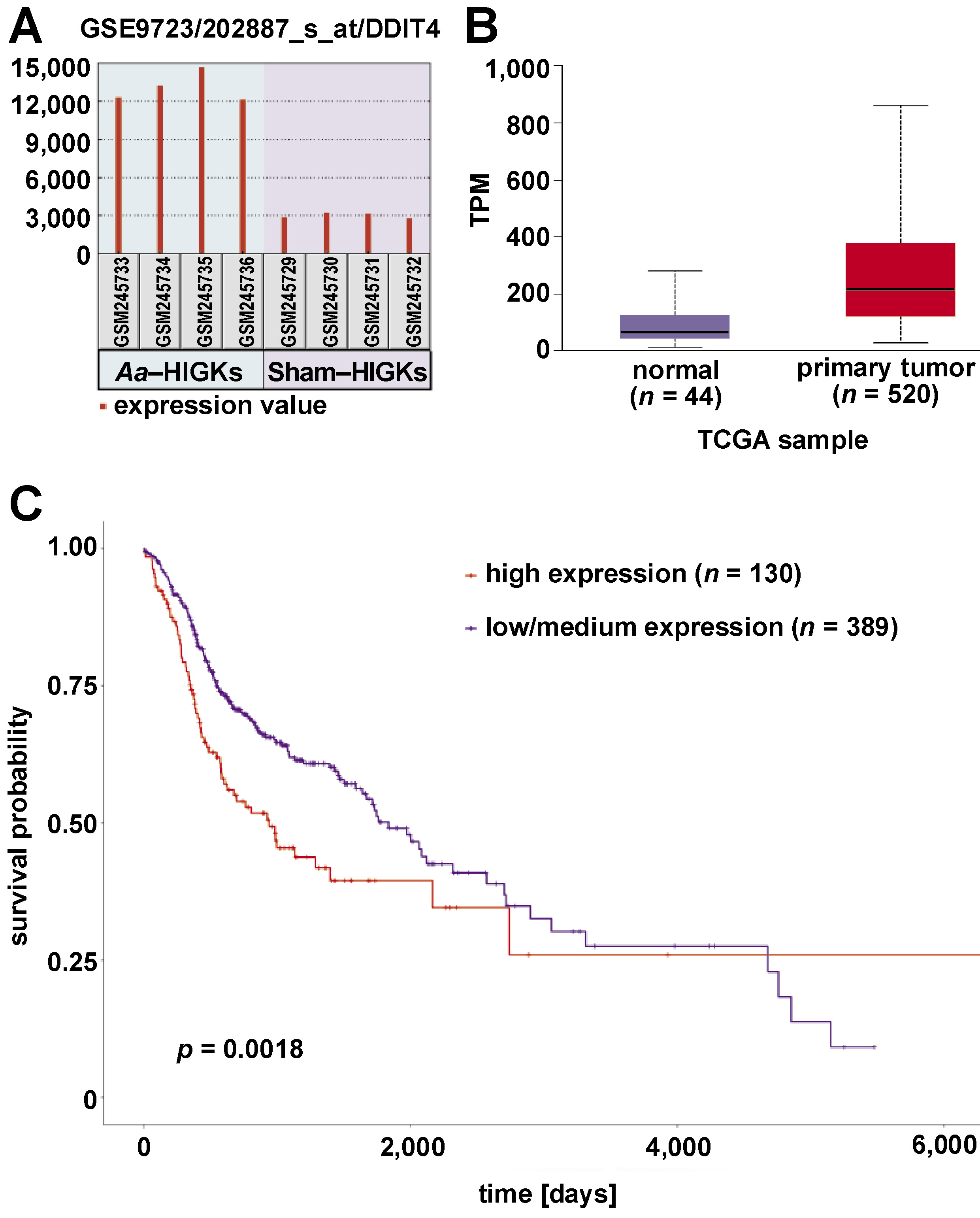
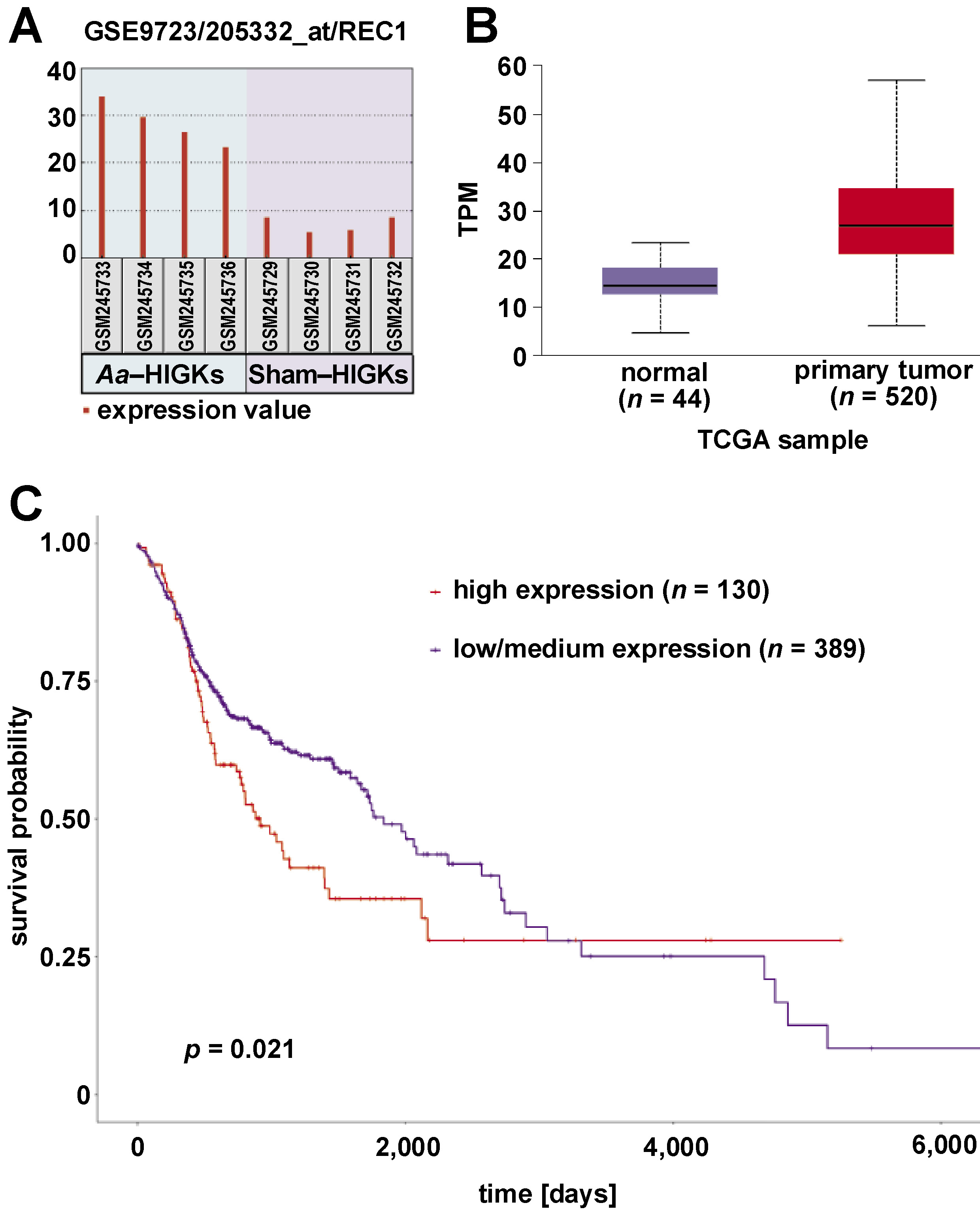
The top 25 genes exhibiting differential gene expression in A. actinomycetemcomitans-infected HIGKs were investigated for their expression profiles in the HNSCC dataset using the UALCAN platform. Genes with similar expression patterns in A. actinomycetemcomitans-infected cells and the HNSCC dataset were selected for further analysis (Table 2). Among the analyzed genes, DDIT4 and RCE1 demonstrated similar expression patterns in A. actinomycetemcomitans-infected HIGKs and the HNSCC dataset (Figure 2, Figure 3). Both DDIT4 (p < 0.001) and RCE1 (p < 0.001) were significantly upregulated in the HNSCC patients and positively correlated with reduced survival. In both cases, increased expression led to poor prognosis in patients (DDIT4: p = 0.0018; RCE1: p = 0.021).
Gene ontology analysis
The gene enrichment analysis of the top 25 DEGs provided insight into the pathways associated with these genes. Although the DEGs did not form clusters, individual genes were correlated with 10 different pathways, of which the Cadherin, Notch, transforming growth factor beta (TGF-β), Toll receptor, and Wnt signaling pathways were linked to carcinogenesis (Figure 4).
Protein–protein interaction analysis
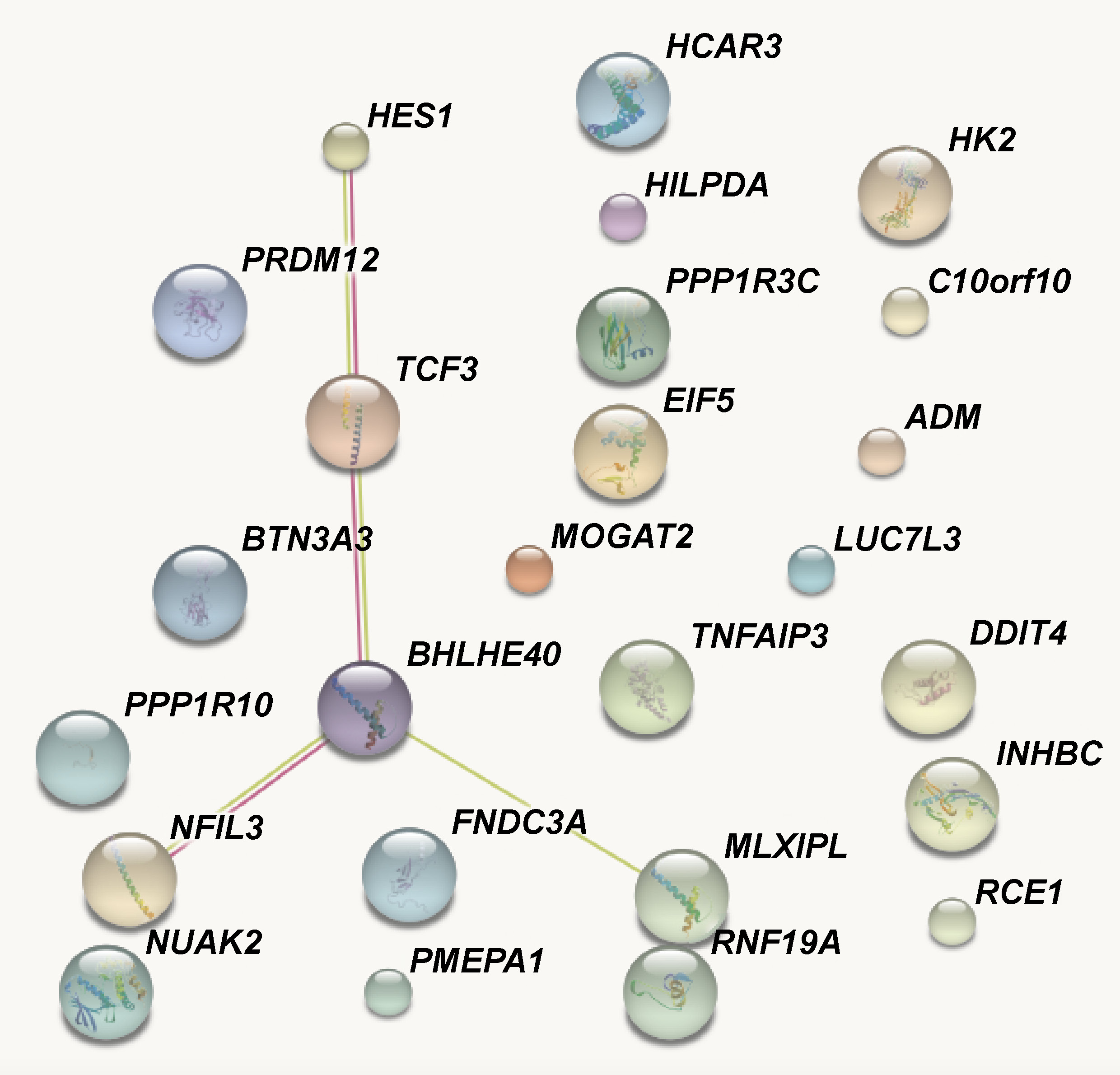
The protein–protein interaction analysis revealed findings similar to those obtained from the gene ontology analysis. Five genes, namely HES1 (hairy and enhancer of split 1), TCF3 (transcription factor 3 (E2A immunoglobulin enhancer binding factors E12/E47)), BHLHE40 (basic helix-loop-helix family member e40), NFIL3 (nuclear factor, IL-3 regulated), and MLXIPL (MLX-interacting protein-like) formed an interaction cluster. The remaining genes appeared as independent units. The text mining process identified an interaction between BHLHE40 and MLXIPL, and the other 3 genes showed experimentally determined interactions (Figure 5).
Discussion
The microbiome, defined as the collection of microorganisms inhabiting the human body, plays a crucial role in maintaining good health. However, dysbiotic shifts in the microbiome have been linked to several diseases, including autoimmune, neurodegenerative and metabolic diseases such as cancer. Interestingly, the microbiome also presents promising opportunities for the identification of microbiome-associated markers that may facilitate the early detection of individuals at high risk of disease development. Recent studies have highlighted the role of the oral microbiome in cancer development, with certain oral bacteria, such as Tannerella forsythia and Porphyromonas gingivalis, playing important roles. By identifying significant differences in the oral microbiome of cancer patients, researchers may improve strategies for early identification of high-risk individuals, improved prevention and treatment.18 A meta-analysis was conducted to systematically assess the impact of periodontal bacterial infections on cancer incidence and prognosis. A total of 39 studies involving 7,184 participants were included in the analysis. The study results indicated a positive association between periodontal bacterial infections and cancer incidence.19 A study by Handfield et al. evaluated the gene expression profile in HIGKs co-infected with P. gingivalis and A. actinomycetemcomitans.13 The gene ontology analysis revealed approx. 55 apoptosis-associated genes, among which TP53 emerged as the major node of the network.13 This scenario mimics the oral environment in which the cells have undergone some dysplastic changes, and the exposure of such cells to bacteria or T cells regulates key pathways associated with carcinogenesis. In this context, the HIGK cell line transfected with the HPV16 E6/E7 gene may serve as an excellent model for studying the effects of bacterial exposure on transformed cells. Notably, the majority of oropharyngeal cancer cases are associated with HPV16.
Recent studies have investigated the influence of oral pathogens on the development and diversity of oral tumors. In one study, oral cancer cells were exposed to different pathogens to better understand this topic.20 The researchers treated the cells with human defensins, which are antimicrobial peptides of the innate immune system, and subsequently analyzed cell proliferation, the expression of oncogenic relevant defensin genes, and epidermal growth factor receptor (EGFR) signaling after stimulation. The results showed that exposure to P. gingivalis in combination with human α-defensins increased cell proliferation, while exposure to A. actinomycetemcomitans led to cell death. However, the outcomes of both pathogens on oncogenic relevant defensin gene expression and EGFR signaling differed. Despite the differences in primary proliferation, both pathogens demonstrated opposite effects on tumor cell proliferation. The study also hypothesized that the antimicrobial peptides could serve as EGFR-binding ligands, thereby regulating the behavior of oral tumor cells.20 A study of 361 pancreatic adenocarcinoma patients and 371 controls demonstrated that oral pathogens (P. gingivalis and A. actinomycetemcomitans) were associated with an increased risk of pancreatic cancer. At the same time, the phylum Fusobacteria and the genus Leptotrichia were linked to a lower risk of disease development. These findings further support the potential role of oral microbiota in the development of pancreatic cancer.21
A comprehensive review by Ungureanu et al. highlighted the complex relationship between periodontal disease and pancreatic cancer.22 Periodontal infections and the associated chronic inflammation have been linked to the development of various systemic conditions, including pancreatic cancer. Recent studies have found that poor oral health, a common manifestation of periodontitis, may increase the risk of pancreatic cancer by 50%, representing a potential risk factor for disease development. Certain oral bacteria, including P. gingivalis and A. actinomycetemcomitans, may elevate the risk of pancreatic cancer, while other beneficial microorganisms, such as Leptotrichia and Fusobacteria, may decrease the risk of the disease. Researchers have observed changes in the oral cavity, gut and pancreatic tissue microbiomes in patients with pancreatic cancer. These observations underscore the importance of maintaining oral health and implementing appropriate periodontal treatment to reduce the incidence of pancreatic cancer.22 Reports from various experimental studies confirm the association between periodontal pathogens and various forms of cancer.
The present study is the first to correlate an array of DEGs identified in A. actinomycetemcomitans-infected HIGKs with the expression pattern in primary tumor samples of HNSCC patients. Among numerous DEGs, the top 25 genes were selected for further investigation. Two of these, namely DDIT4 and RCE1, were significantly upregulated in both datasets. The overexpression of these genes in the HNSCC dataset resulted in a poor prognosis, based on the observed p-values (Figure 2C, Figure 3C). A study conducted by Tajik et al. aimed to investigate the function of DDIT4 in pancreatic tumors and its clinical significance.23 The authors demonstrated elevated expression of DDIT4 in tumor samples, which was directly proportional to the tumor grade and lymphovascular invasion. Immunohistochemical analysis confirmed high DDIT4 protein levels in pancreatic tumors. Nuclear expression was linked to aggressive features, whereas membranous expression was associated with less aggressive behavior in pancreatic ductal adenocarcinoma (PDAC). Although no correlation was found between DDIT4 and patient survival, DDIT4 appeared to be highly sensitive as a diagnostic marker.23
Epithelial–mesenchymal transition (EMT) plays a critical role in oral cancer progression. During EMT, epithelial cells acquire mesenchymal characteristics, promoting invasion and metastasis.24 An experimental study aimed to investigate the role of DDIT4 in cervical cancer metastasis. DDIT4 is a gene that is induced by hypoxia. It is associated with poor prognosis and lymph node metastasis in patients with early-stage cervical cancer. In vitro experiments demonstrated that the reduction in DDIT4 levels inhibited cell migration and invasion, which was linked to reduced expression of EMT-related proteins and decreased activation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway. In addition, DDIT4 was found to promote tumor progression in a mouse model. These results suggest that DDIT4 may serve as a prognostic indicator and promote metastasis in cervical cancer through the EMT and NF-kB pathways.25 The protein–protein interaction and gene ontology analyses conducted in the present study revealed potential connections with the EMT, inflammatory networks and unique pathways related to tumorigenesis, such as Wnt, Notch and TGF-β signaling pathways.
A recent study has shed light on the importance of DDIT4 in the phenotype of triple-negative breast cancer (TNBC). Using RNA-seq and data mining, the authors identified the overexpression of DDIT4, which has been linked to poor prognosis, resistance to neoadjuvant chemotherapy, and a negative impact on the immune microenvironment. Further analysis of DDIT4 and its hub genes (ADM, ENO1, PLOD1, CEBPB) showed associations with apoptosis, cell cycle and EMT pathways, indicating the potential of DDIT4 as a prognostic biomarker and a therapeutic target for improving TNBC treatment strategies.26 These reports are consistent with the observations of the present study and provide further evidence supporting the role of DDIT4 in the development of head and neck cancers.
The other gene identified in the present study, RCE1, encodes the Ras converting CAAX endopeptidase 1, a crucial end-proteolytic enzyme involved in cellular processes, including carcinogenesis. An earlier study investigated the expression of RCE1 in colorectal carcinoma (CRC) tissues and evaluated its prognostic significance. Immunohistochemical analysis of 244 CRC specimens revealed reduced RCE1 expression in tumor tissues compared to adjacent non-tumor tissues. Lower RCE1 expression independently predicted poor overall and disease-free survival. Mechanistically, RCE1 influenced tumor invasion through the p38 pathway, and RCE1 knockdown enhanced the invasiveness of CRC cells. These findings suggested that RCE1 functions as a tumor suppressor in CRC, impacting invasion and predicting unfavorable patient outcomes.27 Another investigation involving 216 hepatocellular carcinoma (HCC) tissues, 216 adjacent non-tumorous tissues and 20 normal liver tissues revealed decreased RCE1 expression in HCC, which correlated with early recurrence markers and low alpha-fetoprotein levels. Survival analysis identified reduced RCE1 expression as a predictor of poor outcomes. Functional studies demonstrated the role of RCE1 in suppressing HCC proliferation, migration and invasion. In contrast, in vivo studies confirmed its impact on tumor growth and metastasis via the P38 signaling pathway and EMT induction.28 Although these studies established the tumor-suppressive function of RCE1, several other publications portrayed the oncogenic functions of RCE1 in other malignancies. A study evaluating RCE1 mRNA and protein expression in prostate cancer (PC) and benign prostatic hyperplasia (BPH) tissues revealed markedly higher levels of the gene. The expression of RCE1 in PC was correlated with clinicopathologic features, including Gleason score, T class and distant metastasis. Immunohistochemical analysis detected RCE1 in 93.24% of PC tissues, and higher RCE1 levels were associated with substantially shorter survival time. Therefore, RCE1 has been established as an independent prognostic biomarker for PC. Similar findings were observed in the present study, emphasizing the need for in-depth investigation of the molecular mechanisms involving RCE1 in head and neck cancer.29 The mechanisms through which A. actinomycetemcomitans induces the expression of these genes remain to be elucidated. In recent studies, epigenetic mechanisms have been linked to the stages of carcinogenesis.30 The pan-genomic modifications in the DNA, such as methylation, histone modification and non-coding RNA-mediated regulation of gene expression, are key drivers of cancer-related pathways. Further investigation of the epigenetic components is warranted to better understand the host–pathogen interactions contributing to disease progression.
Limitations
The present study has several limitations in addition to its strengths. First, the study design was based primarily on computational analyses. Therefore, the findings require validation using experimental approaches and in vivo models. Second, gene expression profiles are influenced by numerous factors, including exposure to carcinogens, lifestyle habits, infectious diseases, inflammatory conditions, age, sex, tumor stage, and ethnic background. Consequently, controlled experimental settings are necessary to establish a definitive association between microbial pathogens and cancer development. Third, although the study demonstrated the relationship between candidate gene expression and patient survival, epigenetic mechanisms may exert additional effects on the prognosis of HNSCC patients, which should be explored further. Finally, the cell line used in the present investigation was an immortalized HIGK line. Although a previous study has reported that HIGKs behave like primary gingival epithelial cells (GECs) under experimental conditions,13 it is worthwhile to replicate these findings using normal fibroblasts to determine potential deviations in gene expression profiles. This experimental design could eliminate the possibility of differential expression due to transfection with viral components that ensure the immortalization of cells.
Conclusions
Microbial dysbiosis and dysplastic changes in host tissues may contribute to the malignant transformation of cells. Although not an independent factor for cancer development, microbial components may directly damage genetic material or indirectly activate several crucial pathways, leading to carcinogenesis. Recent advancements in oral and gut microgenomics have created new opportunities for improving the outcomes of patients with cancer. By integrating the 2 fields, researchers hope to gain a deeper understanding of the complex interactions between the oral and gut microbiota, paving the way for the development of effective therapeutic strategies for cardiovascular, cerebrovascular, metabolic, inflammatory, and autoimmune conditions. The present study is the first of its kind to provide insight into the putative genetic components related to A. actinomycetemcomitans-mediated development of HNSCC.
Ethics approval and consent to participate
Not applicable.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Use of AI and AI-assisted technologies
Not applicable.