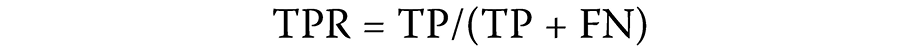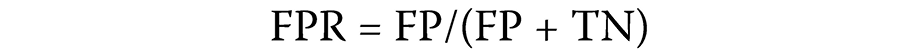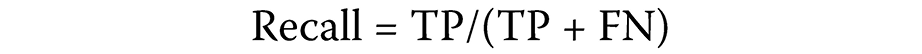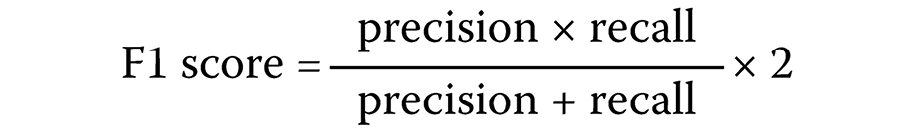
Abstract
Background. The anaerobic commensal Fusobacterium nucleatum is scarce in healthy subgingival dental biofilms but is highly prevalent in periodontal pockets. Numerous genome-wide association studies and gene expression studies using microarrays or RNA sequencing (RNA-Seq) have been performed to better understand the genetic architecture of periodontal disease. However, these investigations have limited predictive capacity for identifying RNAs, particularly non-coding RNAs (ncRNAs). The mechanism of regulation of ncRNAs by F. nucleatum to alter disease progression in mice has not been thoroughly investigated.
Objectives. The aim of the study was to predict previously uncharacterized ncRNAs in F. nucleatum-infected mice using machine learning (ML).
Material and methods. Long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs) were identified from the periodontitis gene expression dataset (GSE225589) obtained from the Gene Expression Omnibus (GEO) database and subsequently preprocessed. Long non-coding RNAs and circRNAs were labeled based on the gene expression. Transcriptomic features were analyzed using 3 ML algorithms: random forest (RF); adaptive boosting (AdaBoost); and naïve Bayes (NB). The dataset was labeled and divided into training (80%) and testing (20%) subsets with cross-validation. Additionally, receiver operating characteristic (ROC) curves, confusion matrices and area under the ROC curve (AUC) values were determined.
Results. The RF and AdaBoost models outperformed the NB model in classifying lncRNAs and circRNAs. Both RF and AdaBoost achieved an AUC of 100%, whereas the NB model achieved a slightly lower AUC of 92%.
Conclusions. This study is the first to apply ML to predict ncRNAs in F. nucleatum-infected mice using transcriptomic data. Random forest and AdaBoost showed superior classification performance in identifying lncRNAs and circRNAs associated with the infection. Further studies with larger cohorts and external validation are needed to confirm these findings.
Keywords: periodontal disease, machine learning, transcriptomics, non-coding RNAs
Introduction
Chronic multifactorial inflammatory periodontitis progressively destroys the tooth-supporting structures and is associated with dysbiotic dental plaque biofilms.1, 2 Approximately 100 different microbial species inhabit the human oral cavity.3, 4 Although most oral bacteria are commensals, a small proportion are hazardous.5, 6 The formation of bacterial biofilms on tooth surfaces is identified as the main cause of periodontal disease.7, 8, 9, 10, 11 Common oral bacteria include Porphyromonas gingivalis, Tannerella forsythia, Prevotella intermedia, Campylobacter rectus, Eikenella corrodens, Fusobacterium nucleatum, Aggregatibacter actinomycetemcomitans, Treponema species, and Eubacterium species.
Oral bacteria produce various polysaccharides and glycoproteins that enable co-aggregation with planktonic microorganisms and adhesion to surfaces (co-adhesion). The acquired pellicle formed on tooth surfaces, composed primarily of salivary glycoproteins and antibodies, facilitates bacterial attachment. Biofilm-associated microorganisms exhibit distinct characteristics compared with planktonic bacteria, including close spatial organization, production of a self-generated extracellular matrix, reduced metabolic activity, and quorum sensing mechanisms that enhance coordinated survival and persistence.
The most prevalent colonizers are gram-positive facultative anaerobes, particularly Streptococcus and Actinomyces species.12 The build-up of dental plaque causes a decrease in oxygen levels, favoring colonization by anaerobic bacteria. Fusobacterium species serve as bridging organisms between primary and secondary colonizers.13 Socransky et al. classified the microorganisms into microbial complexes based on their color.14 The red and orange complexes are strongly associated with periodontal disease in the subgingival region.7, 15 Among these organisms, F. nucleatum is considered a key species in the etiology of periodontitis.
Fusobacterium nucleatum is an anaerobic commensal bacterium present in low concentrations in healthy subgingival biofilms16 but enriched in periodontal pockets.17 It serves as an important bridging organism between early colonizers and periodontal pathogens.18 Fusobacterium nucleatum has been also associated with colorectal cancer, ulcerative colitis, cardiovascular disease,19, 20 and extraoral infections that might be dangerous in pregnancy,19 supporting its classification as an opportunistic pathogen.
Experimental periodontitis models in rats have demonstrated that F. nucleatum infection can cause abscess formation and alveolar bone loss.21 Infection with F. nucleatum and A. actinomycetemcomitans stimulates the production of cytokines such as interleukin (IL)-1. Additionally, tumor necrosis factor (TNF) and IL-17 synergize with IL-1 to enhance the synthesis and expression of additional cytokines (e.g., IL-6), defensins and endothelial activation markers, thereby amplifying the immune response.
Recently discovered non-coding RNA (ncRNAs) include long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs).22, 23 Many diseases are initiated and progress through cis- and trans-regulatory gene expression mechanisms.24, 25, 26 LncRNAs affect the gene regulatory network of host–pathogen interactions and influence biological processes such as cell proliferation, motility, and immune or inflammatory response in cardiovascular, inflammatory and autoimmune diseases.27, 28, 29 The biological functions of circRNAs are not yet fully understood. According to previous studies, circRNAs may function as microRNA (miRNA) sponges, thereby inhibiting miRNA activity, increasing the expression of miRNA target genes, modulating cytokine expression, promoting cell cycle progression, and inhibiting apoptosis.30, 31, 32 Both lncRNAs and circRNAs have been identified as potential diagnostic markers.33, 34
However, in vitro and in vivo studies have investigated F. nucleatum invasion and host response mechanisms.35, 36, 37, 38, 39 Previous research has shown that F. nucleatum infection activates the inflammatory response, induces cytokine production and sends danger signals to human glomerular endothelial cells (GECs).37, 40 The molecular mechanisms underlying immune responses to F. nucleatum infection in mice are unknown. To understand the genetic architecture of this complex trait, several genome-wide association studies and gene expression investigations using microarrays or RNA sequencing (RNA-Seq) have been conducted. The outcomes of these investigations, however, do not provide robust insights. Li and Liang developed LncDC, a Python-based tool that outperformed 6 other algorithms in classifying lncRNAs and mRNAs.41 Using osteosarcoma transcriptomic data, they identified 97 novel lncRNAs, providing potential diagnostic biomarkers or therapy targets.41 However, no studies have investigated how F. nucleatum regulates ncRNAs to influence disease progression in mice. The application of alternative methodologies can improve the generalizability of the results. Machine learning (ML) techniques, combined with resampling strategies, offer a powerful framework for identifying ncRNAs in mice infected with F. nucleatum.
Due to their computational effectiveness in identifying generalizable patterns from high-dimensional datasets generated from small samples, ML techniques have been utilized to analyze high-throughput deep sequencing data.42 Therefore, the current study aimed to apply ML to predict previously uncharacterized ncRNAs in mice infected with F. nucleatum.
Material and methods
Source and processing of data
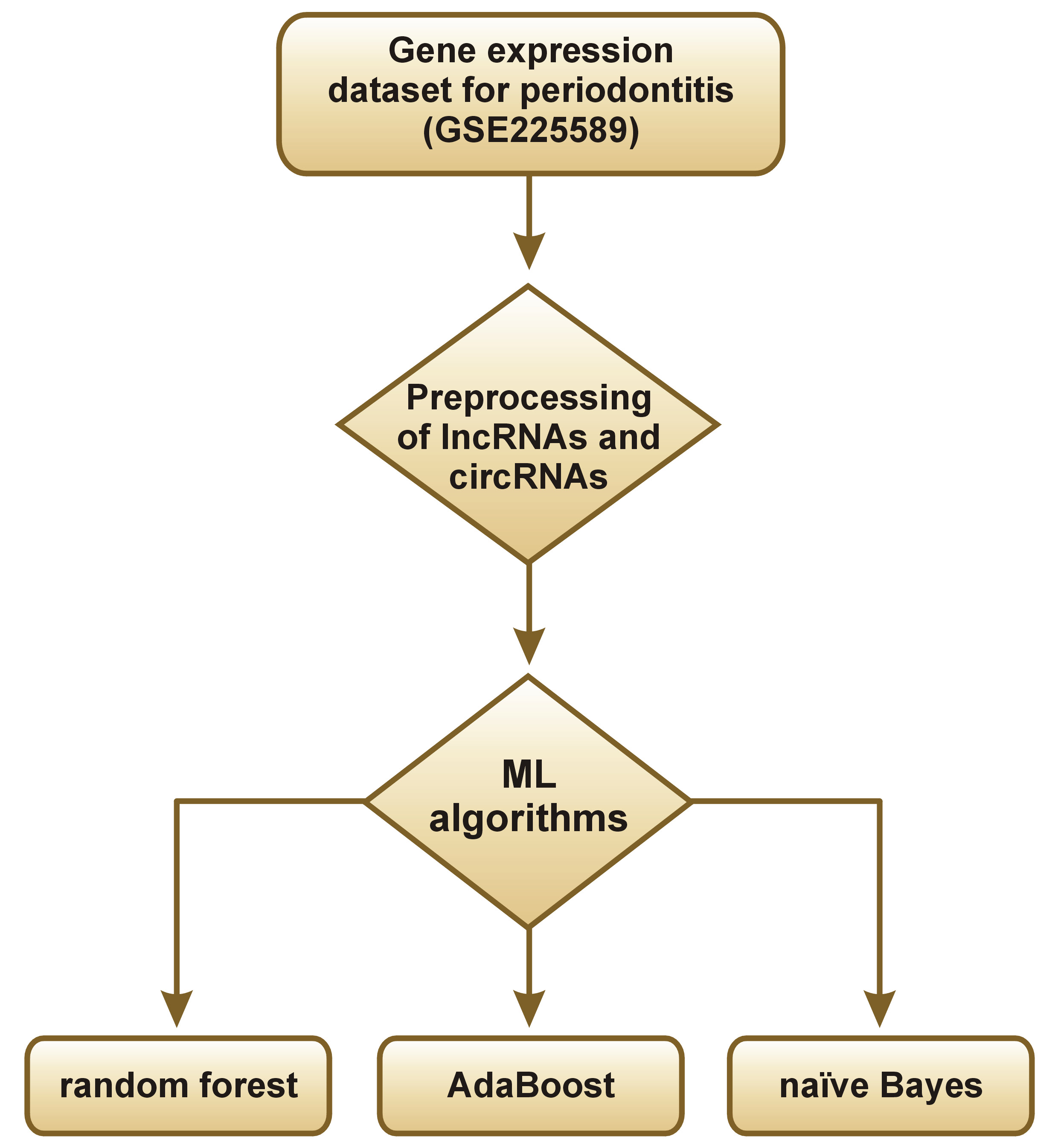
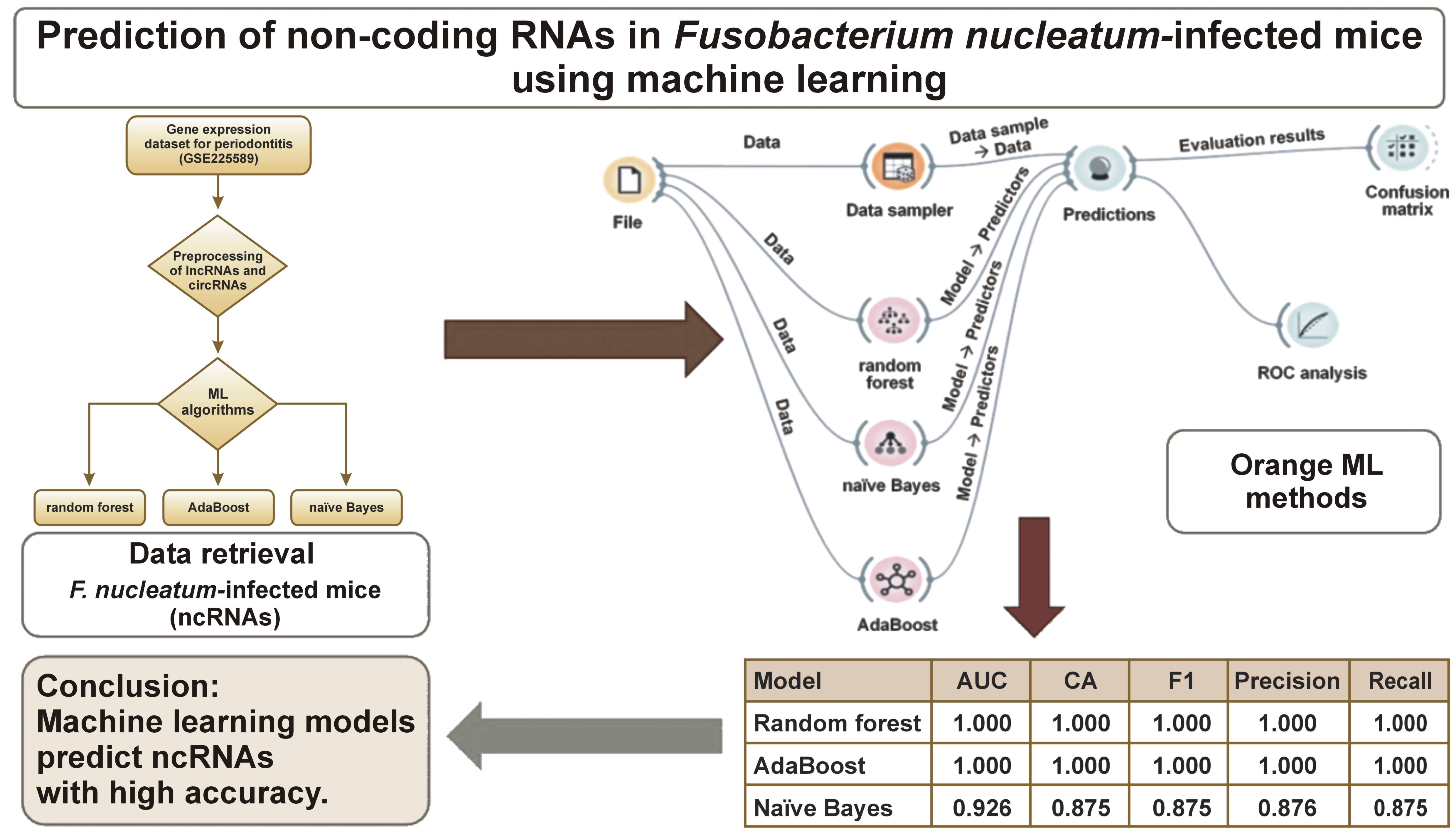
The gene expression dataset for periodontitis (GSE225589) was retrieved from the Gene Expression Omnibus (GEO) database. A comprehensive search of microarray studies was conducted using the keywords “periodontitis” and “Rattus norvegicus”. The epitranscriptomic gene expression data was exported to Microsoft Excel (Microsoft Corp., Redmond, USA), and outliers were removed. The dataset was preprocessed to identify lncRNAs and circRNAs, which were then classified and labeled based on their gene expression features. The processed data was subjected to exploratory data analysis.
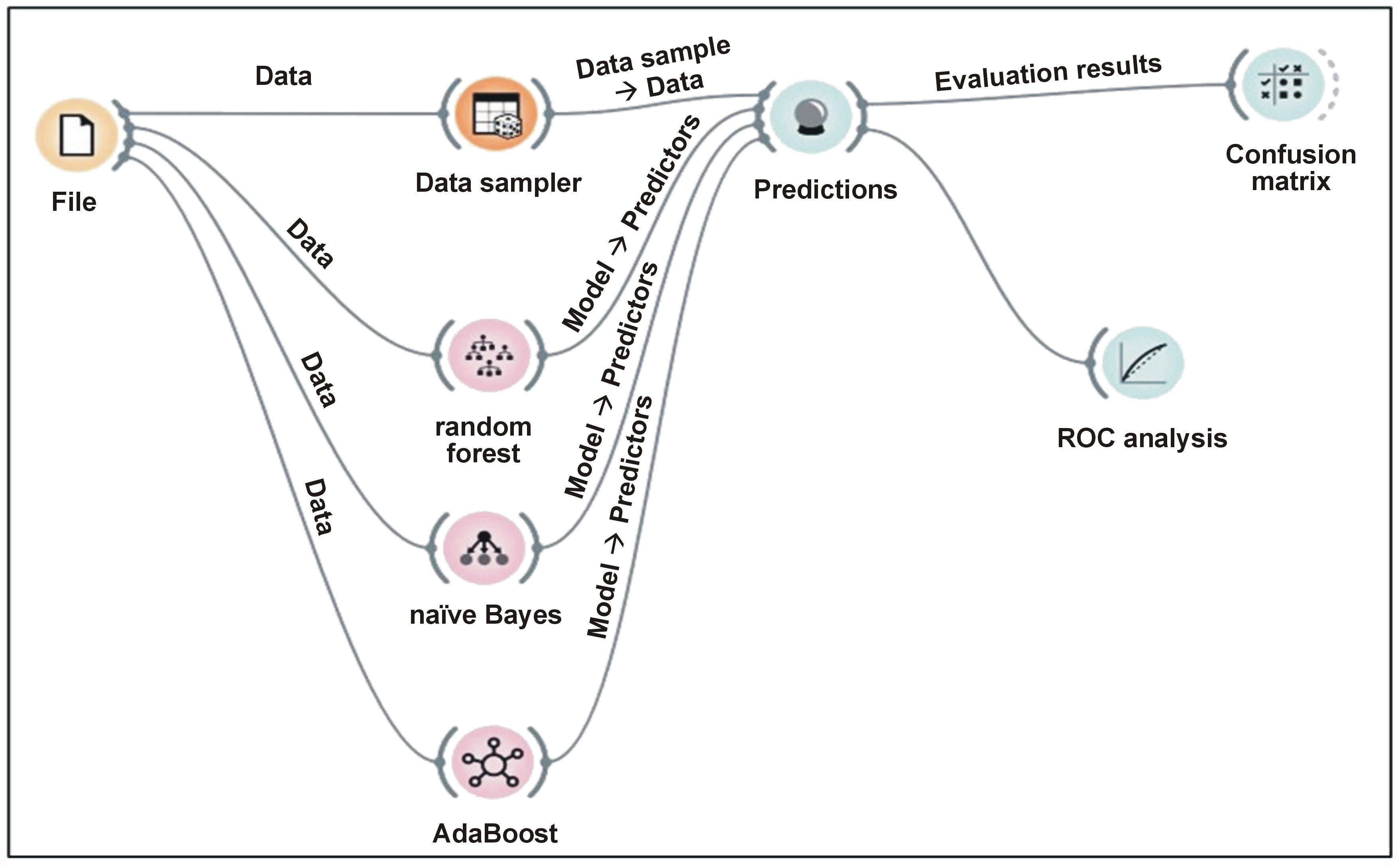
The Orange Data Mining tool (https://orangedatamining.com/download) was used for gene expression analysis, including data upload, transformation, user-interactive visualization, as well as model inference and visualization. These stages involve data input and transformation, interactive visualization, drawing conclusions about data models, and model representation. Data is typically received, processed and visualized by a workflow component, which then generates the analysis for further processing (Figure 1,Figure 2).
Random forest (RF) parameters included up to 100 decision trees, with no strict limitation on tree depth and feature selection. Adaptive boosting (AdaBoost) parameters included the number of boosting iterations and learning rate with several estimators set to 50, and the SAMME (Stagewise Additive Modeling using a Multi-class Exponential loss) learning algorithm. Data balancing techniques, such as oversampling, undersampling and class weighting, were used to address class imbalances in the dataset. Model performance was evaluated using a confusion matrix, receiver operating characteristic (ROC) curve, and area under the ROC curve (AUC). The AUC provides an aggregate performance measure across different classification thresholds, while the confusion matrix summarizes information about the model’s performance in each class. The ROC curve provides a trade-off between sensitivity and specificity, thus facilitating the selection of an appropriate threshold.
Genes with constant expression patterns within the same class were assigned greater weight than genes with erratic expression. This method reduces non-informative genes to improve classification accuracy.
Adaptive boosting
Adaptive boosting is an ensemble learning algorithm that combines weak learners to build a strong classifier. The algorithm fits a classifier to the original dataset and then trains additional classifiers on weighted versions of the data. In each iteration, the weights of misclassified instances are increased, allowing the model to focus on more challenging cases. AdaBoost can be applied to gene expression data from DNA microarray and RNA-Seq platforms for classification tasks.
Random forest
Random forest is a sophisticated ML method used for classification and regression. It constructs numerous decision trees during the training phase. The output of this process is the class, that is, the mode of the classes (classification) or the mean prediction (regression) of the individual trees. Random forest can predict clinical outcomes in gene expression (genes) using profile datasets with several characteristics. The prediction task is a classification problem involving feature representations. The primary objective of this approach is to achieve high classification accuracy. Random forest is effective for the analysis of gene expression datasets, due to its capacity to manage many input variables and their complex interactions. High-dimensional data, such as gene expression profiles, is prone to overfitting; however, this method is robust.
Naïve Bayes
Naïve Bayes (NB) is a basic yet successful ML method that applies Bayes’ theorem with strong (naïve) independence assumptions among features. Due to its simplicity and efficiency, NB is frequently applied to high-dimensional datasets, including gene expression data.
The naïve Bayes classifier (NBC) is widely used in pattern recognition tasks involving gene expression. However, classical estimations of location and scale parameters are sensitive to outliers, which complicates the interpretation of gene expression data using the classical NBC.
Results
The predictive performance of lncRNA and circRNA disease associations was evaluated using stratified 20-fold cross-validation. In each iteration, the dataset was partitioned into training and testing subsets, with most samples used for training and the remainder for testing. During each cycle, similarities between lncRNAs and circRNAs were recalculated based solely on known training associations.
After estimating the association probabilities of the test samples, the samples were arranged according to their association scores. Samples with higher scores were considered more likely to represent true lncRNA–circRNA disease associations. A sample was deemed positive if an observed association existed in the lncRNA–cirRNA disease node pair and its association score exceeded a predefined threshold. The true positive rate (TPR) and the false positive rate (FPR) were calculated as follows (Equation 1 and Equation 2):
where:
FN – number of false negatives (incorrectly classified positive samples);
FP – number of false positives (incorrectly classified negative samples);
TP – number of true positives (correctly classified positive samples);
TN – number of true negatives (correctly classified negative samples).
By varying the threshold, ROC curves were generated. The AUC provided an overall measure of the predictive capability of the model.43
Due to a considerable imbalance between observed lncRNA–disease associations (positive samples) and unobserved associations (negative samples), precision–recall (PR) curves and the area under the PR curve (AUPR) were used to assess predictive performance.44 Precision and recall were defined as follows (Equation 3 and Equation 4):
A precision of 1 indicates that FP = 0. Similarly, a recall of 1 indicates that FN = 0. An ideal classifier achieves precision and recall of 1, corresponding to 0 false positives and false negatives. As the number of false negatives increases, the recall decreases because the denominator (TP + FN) grows relative to TP.
To jointly evaluate precision and recall, the F1 score was calculated (Equation 5):
The AUC–ROC curves and, most importantly, recall, precision, specificity, and accuracy, were derived from the confusion matrix. The target feature in this test consisted of 2 groups of predictors: lncRNAs and circRNAs. In this study, stratified 20-fold cross-validation was applied. The confusion matrix summarizes the number of true positives, true negatives, false positives, and false negatives generated by the model on the test data.
Identification and prediction of non-coding
RNAs and construction of classification models
Twenty-fold cross-validation was used to determine model accuracy and AUC after classification using the ML framework (Figure 2). The outcomes demonstrated that, for lncRNAs and circRNAs, accuracy and AUC value reached 100%. To classify lncRNAs and circRNAs, RF, AdaBoost and NB classification models were built. The RF and AdaBoost models achieved the highest accuracy (AUC = 1.000), whereas the NB model achieved a slightly lower AUC of 0.926 (Table 1). These findings indicate that ML-based classification algorithms targeting lncRNAs and circRNAs demonstrate high diagnostic accuracy.
Prediction and expression of non-coding RNAs
The ROC analysis was conducted to assess the predictive utility of the selected lncRNAs and circRNAs. All targeted ncRNAs demonstrated AUC values greater than 0.7, indicating acceptable predictive performance. These findings suggest that the identified ncRNAs may serve as prognostic markers in mice infected with F. nucleatum. However, the individual AUCs of the selected lncRNAs and circRNAs were lower than the overall classification performance of the RF, AdaBoost and NB models, suggesting that the integrated ML approach provides superior predictive accuracy compared with single biomarkers.
Evaluation of the results using the confusion matrix of the RF model correctly classified 110 lncRNAs as true positives as 114 circRNAs as true negatives (Table 2).
Similar to the RF model, AdaBoost correctly identified 110 lncRNAs as true positives and 114 circRNAs as true negatives (Table 3).
The NB model correctly classified 98 lncRNAs as true positives and 98 circRNAs as true negatives. However, it misclassified 12 lncRNAs as false negatives and 16 circRNAs as false positives (Table 4).
Discussion
Fusobacterium nucleatum is a gram-negative, obligate anaerobic bacillus named for its slender, spindle-shaped morphology.45 It has been associated with the etiology of periodontitis and contributes to dental plaque formation.46 Acting as a bridging organism between commensal bacteria and periodontal pathogens on tooth and epithelial surfaces, F. nucleatum plays a crucial role in mediating physical interactions between gram-positive and gram-negative bacteria.47 The bacterium expresses multiple adhesins that facilitate binding to other microorganisms and cells, thereby enhancing pathogenicity. The most important virulence factor in F. nucleatum is Fusobacterium adhesin A (FadA), an adhesion protein.19 Fusobacterium adhesin A exists as mature FadA (mFadA), a secreted protein of 111 amino acids, and pre-FadA, a 129-amino-acid precursor form.48 The active complex (FadAc), composed of both forms, enables binding and invasion of host cells.48, 49
Machine learning applications in transcriptomics have quickly expanded, enabling computational analysis of gene expression data generated by techniques such as RNA-Seq. Using ML approaches, researchers can detect differentially expressed genes, categorize samples into groups, predict gene functions, and find hidden molecular patterns. These capabilities provide novel biological insights and support biomarker discovery. A variety of ML methods, including decision trees, support vector machines, RFs, and deep learning models, have been applied to address the high dimensionality and complexity of transcriptomic datasets. Such strategies have great potential to contribute to our understanding of disease diagnosis, gene regulation, and the development of personalized medicine.50 Because experimental biological research is often time-consuming and costly, computational prediction of disease-associated lncRNAs via bioinformatics has become increasingly common. In recent years, many lncRNA–disease association prediction (LDAP) models have been proposed, including models based on biological networks, models independent of known lncRNA–disease relationships, and ML-based frameworks.
Non-coding RNAs are RNA molecules that do not encode proteins but play significant regulatory functions in several biological processes. Once considered transcriptional noise, ncRNAs are now recognized due to their numerous applications and influence on gene expression.
Well-established subclasses of RNAs include transfer RNAs (tRNAs), which are essential for protein synthesis by transporting amino acids to the ribosomes, and ribosomal RNAs (rRNAs), which form the structural core of ribosomes involved in protein synthesis. Extensive research has been conducted on these RNAs due to their pivotal role in the fundamental functioning of cells. In addition, messenger RNAs (mRNAs) can bind to short RNA molecules, called miRNAs, which can either promote or hinder the translation of mRNAs. MicroRNAs regulate gene expression and play a role in a variety of cellular functions, such as differentiation, development and disease progression.
Another well-known family of ncRNAs is lncRNAs. These larger RNA molecules are transcribed from the genome, yet they do not encode proteins. LncRNAs are involved in several regulatory processes, including post-transcriptional processing, chromatin remodeling and transcriptional control. Additionally, they have been linked to crucial biological processes such as cellular differentiation, the etiology of disease and embryonic development.
Recent studies have also highlighted the functions of enhancer RNAs (eRNAs) and circRNAs in gene regulation. CircRNAs are covalently closed RNA molecules produced during transcription by a back-splicing process. These cells can affect the expression of genes through interactions with RNA-binding proteins or by acting as miRNA sponges. While eRNAs are expected to facilitate enhancer–promoter interactions and modulate gene transcription, they are translated from enhancer regions of the genome.
The functional roles and regulatory mechanisms of ncRNAs are the subject of ongoing research. Dysregulation of ncRNAs has been associated with numerous diseases, including cancer, neurological disorders, periodontal disease, and cardiovascular conditions. The potential of ncRNAs in diagnostics and therapeutics could be utilized in the development of novel biomarkers and focused therapies.
In the present study, we aimed to predict ncRNAs associated with F. nucleatum infection in mice to better understand the etiology of F. nucleatum-related disorders. Orange, an open-source data visualization and ML toolkit developed in Python, has been used to build and analyze predictive models. The tool supports popular algorithms like RF, neural networks, AdaBoost, NB, and logistic regression, but in our study, 3 models were used: RF; AdaBoost; and NB.
The gene expression dataset for periodontitis (GSE225589) was obtained from the GEO database. Using GEO2R, 250 differentially expressed genes (DEGs) were identified by comparison with appropriate controls. The 3 ML algorithms, namely RF, AdaBoost and NB, were evaluated for accuracy. The RF and AdaBoost models achieved superior and consistent accuracy compared with the NB model. Specifically, RF achieved an AUC of 1.000, classification accuracy (CA) of 1.000, F1 score of 1.000, precision of 1.000, recall of 1.000, and Matthews correlation coefficient (MCC) of 1.000. AdaBoost demonstrated identical performance metrics. In contrast, the NB model achieved an AUC value of 0.926, CA of 0.875, F1 score of 0.875, precision of 0.876, recall of 0.875, and MCC of 0.751.
Integrating proteomics or metabolomics data could further enhance understanding of disease mechanisms and improve predictive performance. Machine learning-based identification of indicators or pathways of periodontitis may facilitate the development of targeted therapeutic strategies or diagnostic tools.51
Several limitations of the present study should be acknowledged, including the relatively small sample size, reliance on a single dataset, and the lack of external validation. The identified DEGs and predictive models require replication in independent datasets to confirm accuracy. Understanding the biological significance and mechanisms of these genes is crucial for further insights. Future research should incorporate multi-omics integration, longitudinal analyses, functional experiments, assessment of potential confounding factors, and development of clinically applicable predictive models. These approaches could provide a more comprehensive understanding of the molecular mechanisms underlying periodontitis, identify novel biomarkers, examine gene expression changes over time, validate the biological relevance of DEGs, and improve patient management and outcomes.
A previous study investigating oral colonization of mice with P. gingivalis, Treponema denticola and T. forsythia demonstrated enhanced intrabony defects and alveolar bone resorption (ABR) during polymicrobial infection.47 These pathogens successfully established oral colonization and induced ABR. Another research demonstrated that chronic oral infection with F. nucleatum has been shown to induce symptoms of periodontal disease in mice, spread via hematogenous routes, alter the host immune system and periodontal risk factors, and cause both pro- and anti-inflammatory reactions.49 According to earlier in vitro studies, the significantly elevated levels of immunoglobulin G (IgG) and IgM after chronic infection suggest that F. nucleatum functions as a powerful B-cell mitogen.52 Additionally, the mitogenic activity has been attributed to the Toll-like receptor 2 (TLR2) adjuvant outer membrane porin FomA.53
The endogenous retroviral-associated adenocarcinoma lncRNA (EVADR) and keratin-7 antisense RNA (KRT7-AS) were among 43 upregulated lncRNAs identified in a recent transcriptome investigation of F. nucleatum-infected colon cancer cells.54 Fusobacterium nucleatum has been reported to promote the progression of oral squamous cell carcinoma (OSCC).54 A lncRNA, MIR4435-2HG-5p, has been shown to be upregulated in F. nucleatum-infected OSCC cells, where it functions as a miRNA-296-5p sponge, activates AKT2 signaling, and contributes to AKT2-induced carcinogenesis.55, 56, 57, 58, 59
The role of lncRNAs in F. nucleatum-infected mice sheds light on the complex regulatory mechanisms underlying diseases associated with this pathogen. Previous studies suggest that F. nucleatum infection can alter host lncRNA expression, which may be crucial in regulating host immune responses, promoting bacterial survival and accelerating disease progression. One of the main functions of lncRNAs in infected mice is the control of host immune responses. LncRNAs can function as molecular scaffolds or ploys, interacting with proteins or other RNA molecules to regulate signaling pathways and gene expression involved in immune control. Additionally, they can control the expression of pro- and anti-inflammatory genes, ultimately affecting the host’s ability to mount an effective immune response against F. nucleatum.
A recent study used mono- and dinucleotide sequences to predict ncRNA regulatory functions.60 A back propagation (BP) neural network with principal component analysis and the Levenberg–Marquardt algorithm trained ncRNAs with accuracies of 81.3% for mixed bacterial ncRNAs and 93.3% for prokaryotic tRNAs.60 Another study trained a classifier (MncR) using RNAcentral data and reported over 97% accuracy in classifying ncRNA classes.61 More recently, ML methods such as logistic regression, RF, eXtreme Gradient Boosting (XGBoost), and decision trees have been employed to distinguish coding from non-coding transcripts and classify ncRNAs. In human datasets, RF achieved accuracies exceeding 83%. Our study obtained similar and highly accurate results.51
Additionally, lncRNAs may influence cellular functions such as cell division, apoptosis and epithelial–mesenchymal transition, all of which are significant in F. nucleatum-associated disorders. Dysregulation of specific lncRNAs in infected mice may promote aberrant cell behavior, potentially contributing to tumor growth, invasion or metastasis in malignancies associated with F. nucleatum. Furthermore, lncRNAs may participate in the direct communication between host cells and F. nucleatum. According to recent research, bacterial lncRNAs may be transferred to host cells, where they can modulate the expression of host genes and signaling pathways. These lncRNAs may play a role in the establishment and persistence of F. nucleatum infection by interfering with host cellular functions.
To fully elucidate the predicted roles of lncRNAs in F. nucleatum-infected mice, further studies using experimental and computational methods are necessary. Integrating transcriptomic analysis with functional assays, including knockdown or overexpression studies, will help clarify the involvement of particular lncRNAs to immune modulation, bacterial persistence and disease development. Additionally, integrative methodologies, such as network analysis and ML, can facilitate the identification of key lncRNA–mRNA interaction networks and improve understanding of the complex regulatory networks.
Investigating lncRNA-mediated regulatory mechanisms in F. nucleatum infection enhances our understanding of host–pathogen interactions and identifies potential therapeutic targets aimed at regulating immune responses, limiting bacterial survival or delaying the onset of disease in F. nucleatum-related conditions.
Conclusions
This study represents the first application of ML approaches to predict ncRNAs in a mouse model infected with F. nucleatum. The superior performance of the RF and AdaBoost models highlights the robustness of ensemble learning for transcriptomic classification. The identified lncRNAs and circRNAs may serve as promising candidates for biomarker development and for advancing our understanding of host–pathogen regulatory mechanisms. Nevertheless, validation in larger and independent datasets, along with functional experimental studies, is essential to confirm their biological and clinical relevance.
Ethics approval and consent to participate
Not applicable.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Use of AI and AI-assisted technologies
Not applicable.