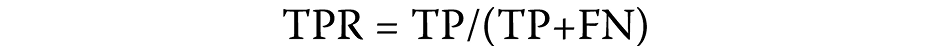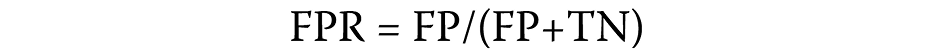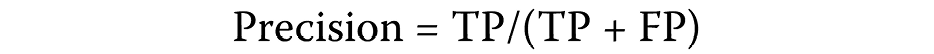Abstract
Background. Oral infections that cause inflammation typically affect the gingival tissues. The immune-inflammatory reactions significantly influence the patient’s vulnerability to periodontal diseases. Numerous studies have found a correlation between persistent inflammation and an increased risk of developing cancer in the afflicted oral epithelium. New research demonstrates a startling connection between periodontal conditions and various forms of cancer, including oral cancer.
Objectives. The aim of the study was to use bioinformatics techniques in order to predict interatomic hub genes in oral cancer and periodontitis.
Material and methods. The datasets were screened for differentially expressed genes (DEGs) in periodontitis and oral cancer using the Gene Expression Omnibus (GEO) database, a gene expression data analysis tool. GeneMANIA was used to identify hub genes between oral cancer and periodontitis. Orange machine learning was conducted for hub gene prediction using random forest, decision tree, AdaBoost, and neural network.
Results. The top 5 hub genes (RSPO4, CDHR2, DDAH2, HLA-J, and IRF3) were prioritized to explore their relationship with oral cancer and periodontal disease. The receiver operating characteristic (ROC) curve was constructed, with the area under the curve (AUC) for random forest at 0.999, for the decision tree at 0.998, for AdaBoost at 1.000, and for the neural network model at 0.865. The AdaBoost model, followed by random forest and decision tree, exhibited the highest level of accuracy (1.000). These results suggest that the 3 models demonstrate good predictability and may facilitate the early detection of periodontitis and oral cancer.
Conclusions. The insights derived from this study may contribute to the development of novel diagnostic and therapeutic techniques for chronic inflammatory periodontitis and oral cancer by utilizing computational approaches and integrating multi-omics data. The identification of interactome hub genes in these diseases has important clinical ramifications. The obtained outcomes may help decipher disease pathways, promote early detection, and create targeted treatments for better patient outcomes. The accurate prediction of hub genes may promote their utilization as biomarkers in the development of individualized treatment plans for both illnesses.
Keywords: hub genes, periodontitis, chronic inflammation, oral cancer, interactome
Introduction
Periodontal infections trigger a prolonged immune-inflammatory response that damages oral tissues locally and encourages chronic systemic inflammation, which contributes to cancer etiology.1 According to the most recent meta-analysis, periodontal disease increases the chance of developing pancreatic, lung, and head and neck malignancies. A recent cohort study with a 10-year follow-up found a substantial link between periodontitis and cancer mortality.2, 3, 4
Oral carcinogenesis is a multifactorial, intricate process that occurs when several genetic changes impact epithelial cells. A few normal keratinocytes undergo a transformation, initiating the process of oral carcinogenesis. Modifications to cytogenetic and epigenetic processes may influence this shift. These alterations can impact the cell cycle, DNA repair procedures and cell differentiation.5 When risk factors are combined with these changes, an unstable keratinocyte develops, eventually evolving into a pre-cancerization field and giving rise to malignant neoplastic alterations.
It is estimated that chronic inflammation is the underlying cause of 15–20% of cancers. Based on the 2017 classification,6 the risk of developing oral cancer has been significantly and independently associated with periodontitis. Studies have also shown a clear causal relationship between the amount and severity of periodontitis,6 as well as between the amount and severity of chronic periodontitis and the risk of oral cancer, even after controlling for common confounding factors, such as smoking, alcohol use and the human papillomavirus (HPV).7, 8, 9, 10, 11 According to recent research, the loss of teeth due to bone loss in periodontitis is a distinct risk factor for head and neck cancer. Additionally, patients with periodontitis exhibit elevated levels of human telomerase reverse transcriptase, whose expression is particularly specific to cancer cells.12, 13 These findings provide substantial evidence to support a strong connection between periodontitis and oral cancer.14, 15, 16, 17, 18, 19, 20, 21, 22, 23 The available studies suggest a potential link between HPV infection, periodontitis and certain types of cancer. Human papillomavirus is a sexually transmitted infection that can cause various types of cancer, including cervical, anal, and head and neck cancers.
The etiology of both periodontitis and oral cancer is multifactorial, involving complex interactions between genetic and environmental factors. The identification of key genes and their interactions in these diseases can provide valuable insights into their pathogenesis and potential therapeutic targets. In recent years, network-based techniques have emerged as effective tools for understanding complex disorders, facilitating the identification of hub genes within protein–protein interaction (PPI) networks. Changes in gene expression frequently signal changes in physiological processes or the onset of disease. The development of diseases, however, may be linked to the molecular functions, biological processes and signaling pathways present in the genetically encoded products.24, 25, 26, 27, 28, 29 Recently, the interactome analysis has gained popularity as a way to understand biological systems’ complicated molecular networks. A molecular interaction network among a cell, tissue or organism is called an interactome. The molecular interactions between proteins, nucleic acids and tiny molecules govern biological processes. Hub genes are essential to the interactome, and they interact extensively with other molecules within the interactome network. Through a network of connections, hub genes control many cellular functions. These genes reveal the functional architecture of cellular pathways and the role of certain genes in biological processes.
Recent clinical and experimental research has focused on the identification of diagnostic indicators. A diagnostic cancer marker may be age-, stage-, tissue-, relapse-, or follow-up-specific and may manifest at any stage of the disease.30 The examination of hub genes31, 32 facilitates the identification of biological pathways associated with the disease, enhancing the specificity of therapeutic options for the condition. In the present study, the Gene Expression Omnibus (GEO) database was examined to obtain an optimal dataset, identify key genes, and determine appropriate directions for future investigations. This research uses network analysis techniques to predict interactome hub genes in periodontitis and oral cancer.
Material and methods
Gene expression databases
The present study used gene expression databases, including GEO33 (periodontitis: GSE186882 and oral cancer: GSE145272; http://www.ncbi.nlm.nih.gov/geo), to examine the expression patterns of the identified genes in periodontitis and oral cancer. These databases provide valuable information about gene expression levels and can help identify genes with consistent and differential expression.
Differential expression analysis
A differential expression analysis was performed using statistical techniques. This analysis compares the expression levels of genes associated with periodontitis and oral cancer, identifying those that demonstrate significant changes in expression.
Network analysis
A gene co-expression network was constructed using GeneMANIA Cytoscape (https://apps.cytoscape.org/apps/genemania).34, 35, 36 Network analysis can facilitate the identification of hub genes based on their connectivity and interactions with other genes within the network.
Analysis of genes with differentially expressed functions
The research on the major differentially expressed genes (DEGs), Gene Ontology (GO) enrichment, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment was carried out using Enrichr (https://maayanlab.cloud/Enrichr).37, 38, 39 Genesets with a false detection rate (FDR) ≤0.05 were considered significantly enriched. A term with a p-value of ≤0.05 and at least 3 genes in its count was deemed significant. After the relevant phrases were grouped into clusters based on commonalities in their membership, the statistically most significant phrase within each cluster was selected to represent the cluster.
Orange machine learning
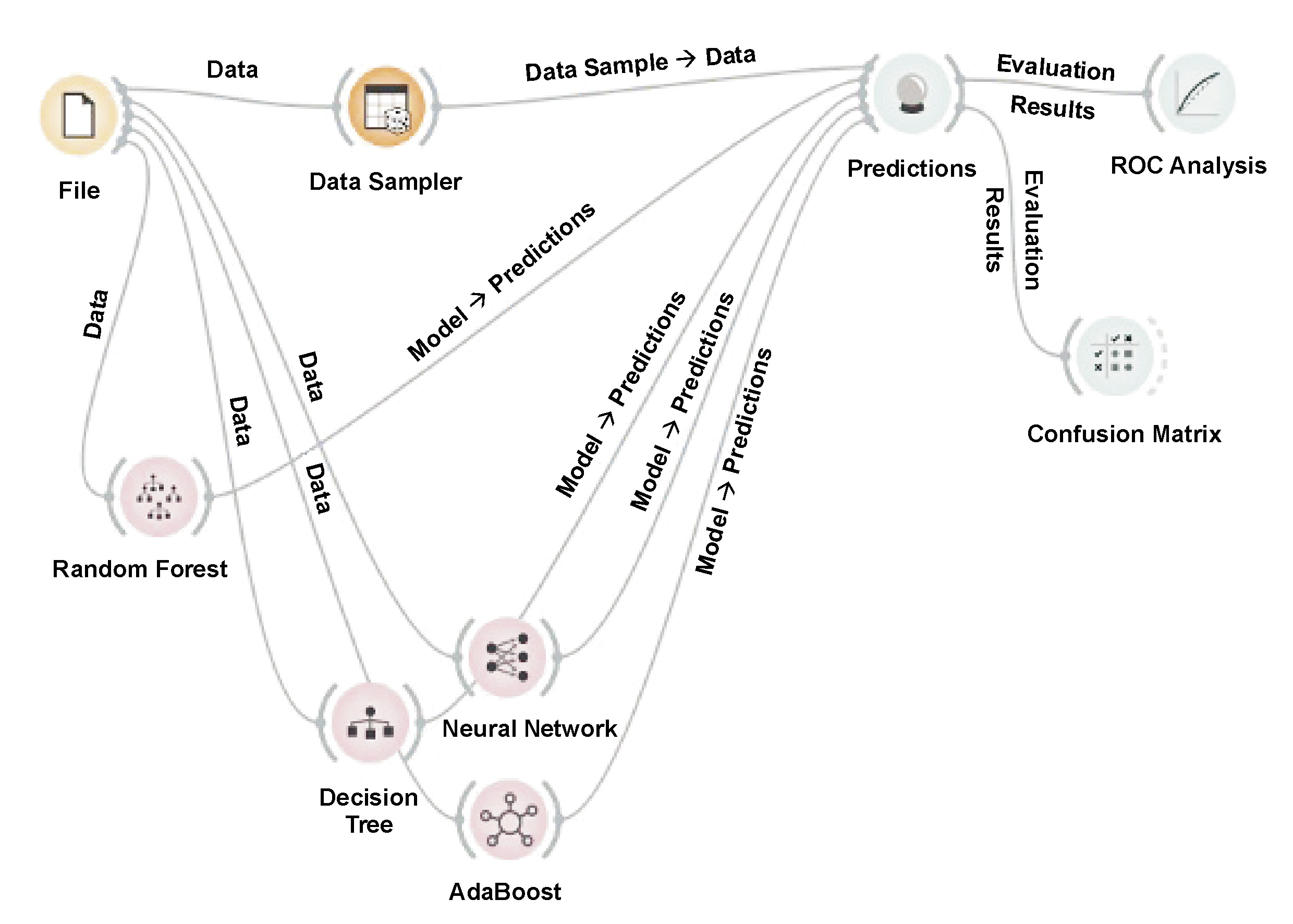
Orange (https://orangedatamining.com) is an open-source toolkit for data visualization and machine learning.40 It provides a user-friendly visual programming interface for data analysis, predictive modeling and visualization. Orange streamlines the machine learning process for users of all skill levels by employing various algorithms and data pre-treatment techniques. Orange is a flexible tool for data analysis and predictive modeling activities, as it facilitates the integration with other well-known machine learning libraries (Figure 1).
The data was split into training and testing portions. Various machine learning algorithms, namely random forest, decision tree, AdaBoost, and neural network were applied for training.
Cross-validation, model scoring and multi-dimensional scaling are all used within the machine-learning workflows implemented in Orange. Orange40 integrates widely used Python libraries for data manipulation and machine learning, including NumPy (http://www.numpy.org), SciPy (https://scipy.org) and scikit-learn (https://scikit-learn.org), and encapsulates their functionality within workflow-based building blocks that provide an interface for adjusting machine-learning parameters or browsing results and associated visualizations of inferred models.
Random forest
Random forest is a supervised machine learning algorithm. It can be used for machine learning problems involving both regression and classification. The system learns and specifies the output based on the majority votes of the individual decision trees. Random forest is the most frequently used method for patient classification and biomarker analysis.
Decision tree
Decision trees are a non-parametric supervised learning method for classification and regression. The tree structure is hierarchical, comprising roots, branches, and internal and leaf nodes.
Adaptive boosting
Machine learning ensemble methods use the boosting technique known as the AdaBoost algorithm, sometimes referred to as Adaptive Boosting. The weights are redistributed to each instance, with heavier weights assigned to instances recognized incorrectly, thus explaining the name.
Neural network
Artificial neural networks (ANNs) are modeled after information processing capabilities of organic nervous systems. These networks consist of interconnected neurons that process information and collaborate to solve specific issues.
Results
Differentially expressed genes in periodontitis and oral cancer
Gene expression datasets for periodontitis (GSE186882) and oral cancer (GSE145272) were selected from the GEO database. A total of 500 DEGs were identified using the GEO2R tool from a dataset concerning periodontitis and oral cancer. The cut-off criteria used to define DEGs were |log2 fold change (FC)| >0 and a p-value of 0.05. GEO2R is a tool used for the analysis of gene expression data from microarray experiments. It helps researchers identify DEGs between different experimental conditions or groups.
GEO2R uses t-tests to compare the means of gene expression levels between different groups or conditions. It calculates the t-value and p-value for each gene, indicating the significance of differential expression. Additionally, the tool calculates the FC values to determine the magnitude of gene expression differences between groups. The fold change is defined as the ratio of gene expression levels between 2 conditions. These statistical techniques help researchers identify significant DEGs, providing insights into biological processes and potential biomarkers.
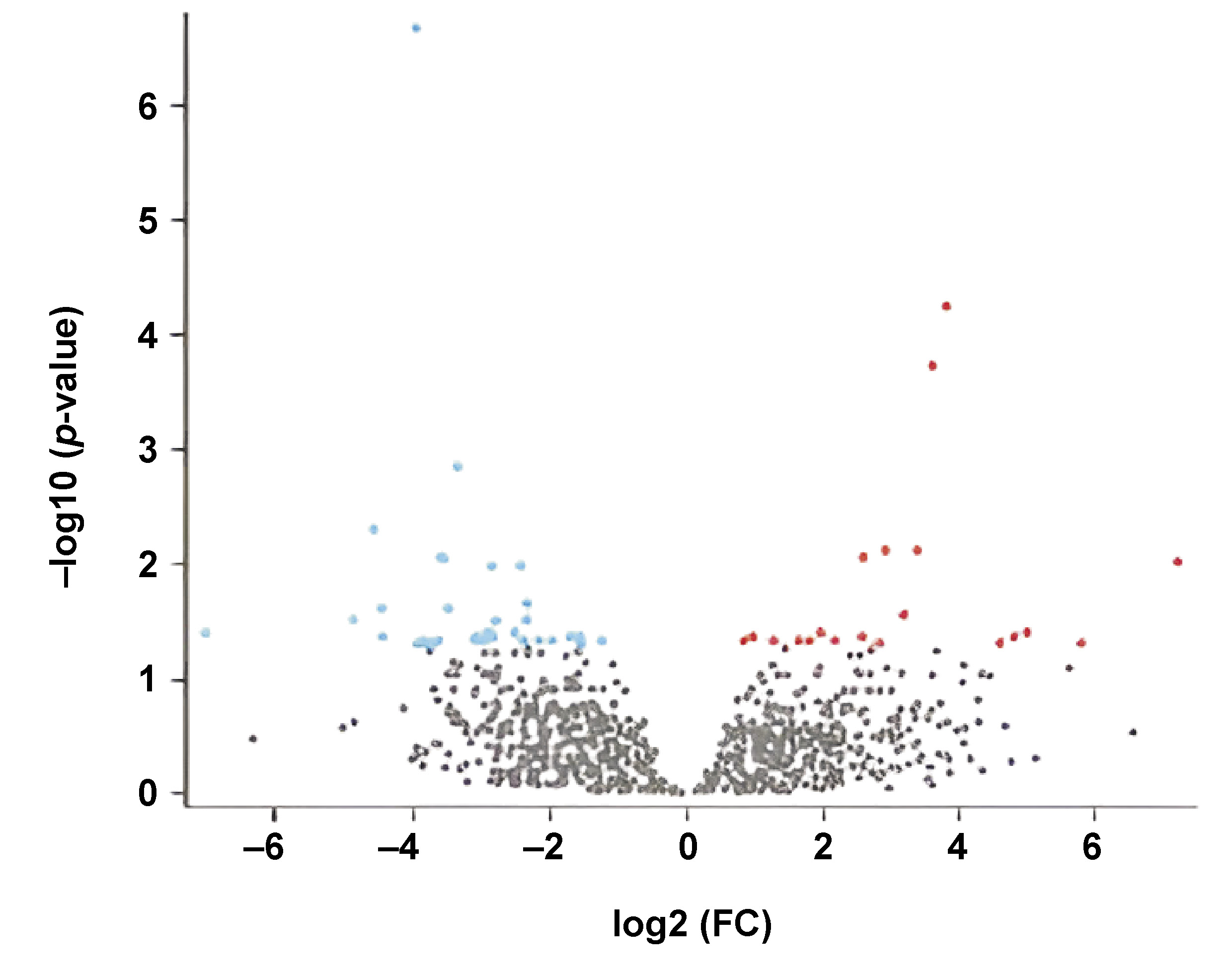
Figure 2 depicts a volcano plot of differential gene expression of oral cancer and periodontitis, with red dots representing upregulated genes, blue dots depicting downregulated genes, and gray dots denoting those without substantial differences.
A total of 250 periodontitis genes and 250 genes associated with oral cancer were identified. The top 5 hub genes (RSPO4, CDHR2, DDAH2, HLA-J, and IRF3) were selected for further study in the context of oral cancer and periodontal disease.
Functional enrichment analysis of DEGs
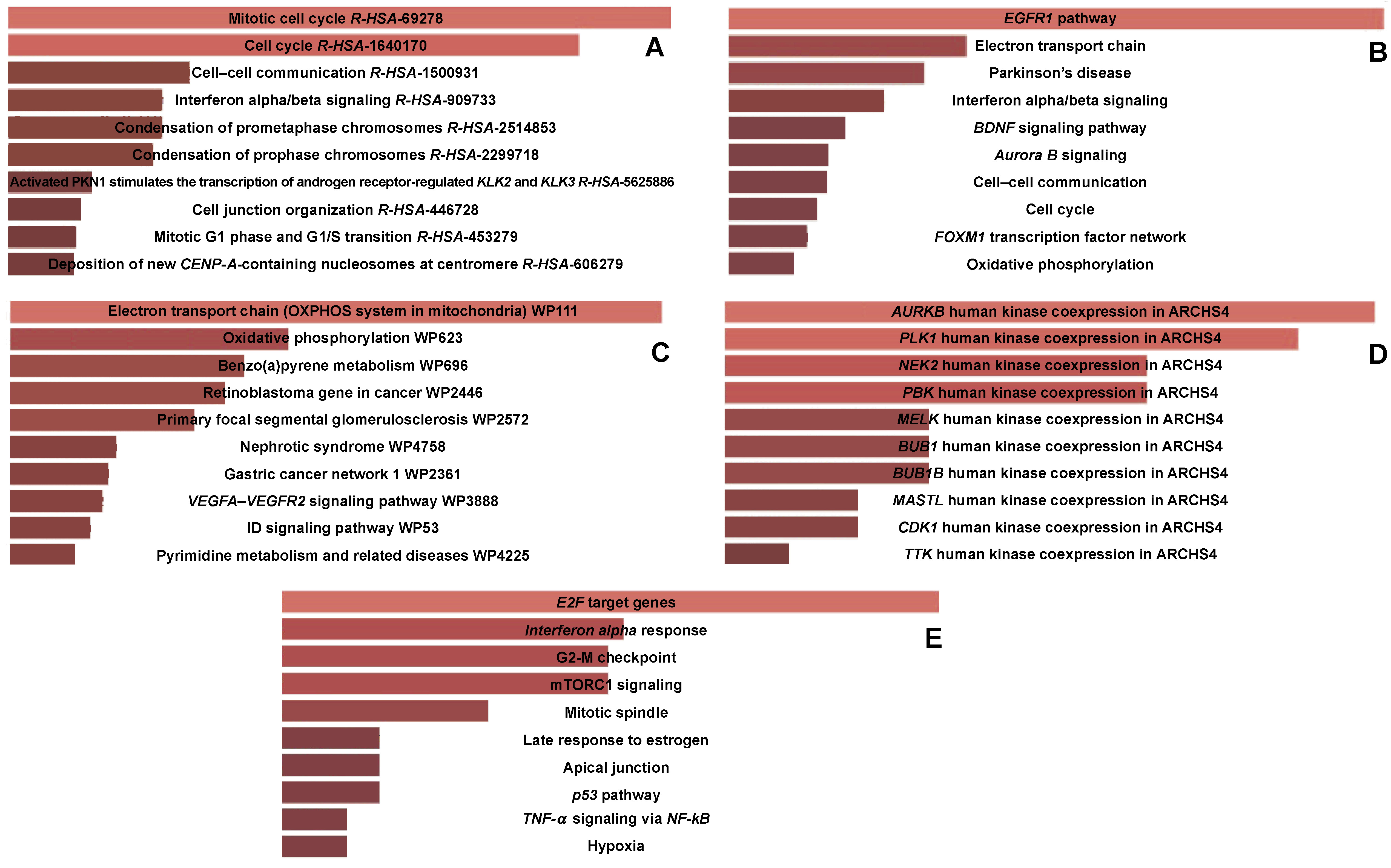
To further leverage the functionalities of DEGs, gene enrichment analysis was performed. Gene ontology enrichment was carried out using Enrichr. According to the analysis, DEGs were more prevalent in immune-related biological processes, including neutrophil degranulation, neutrophil activation implicated in immune response, and neutrophil-mediated immunity (Figure 3). The cell cycle mitotic route, epidermal growth factor receptor (EGFR) signaling pathway, transport chain signaling pathway, aurora kinase B signaling pathway, and tumor necrosis factor alpha (TNF-α) signaling via the nuclear factor kappa B (NF-κB) signaling pathway are among the immune-related pathways that comprise the majority of DEGs (Figure 4A–E). This analysis identified the top 20 clusters with significantly enriched DEGs (Figure 5).
Predictive modeling of hub genes using machine learning
Using the remaining 20% of the testing samples and 80% of the trained samples that were randomly selected for each cross-validation as the training data, we recalculated the similarities between hub and non-hub genes based on the established correlations to evaluate the effectiveness of predicting interactome hub and non-hub genes in periodontitis and oral cancer.
The sample is regarded as affirmative if a hub gene–disease node pair sample demonstrates an observable association and the association score surpasses the established threshold. The true positive rates (TPRs) and false positive rates (FPRs) are primarily calculated to construct a receiver operating characteristic (ROC) curve. The true positive rate is calculated as follows (Equation 1):
where:
TP – true positive: samples that were successfully identified and designated as positive;
FN – false negative: the number of hub genes that were incorrectly classified as not belonging to the positive class.
The false positive rate is quantified using the following formula (Equation 2):
where:
TN – true negative: the number of hub genes correctly identified as not belonging to the positive class.
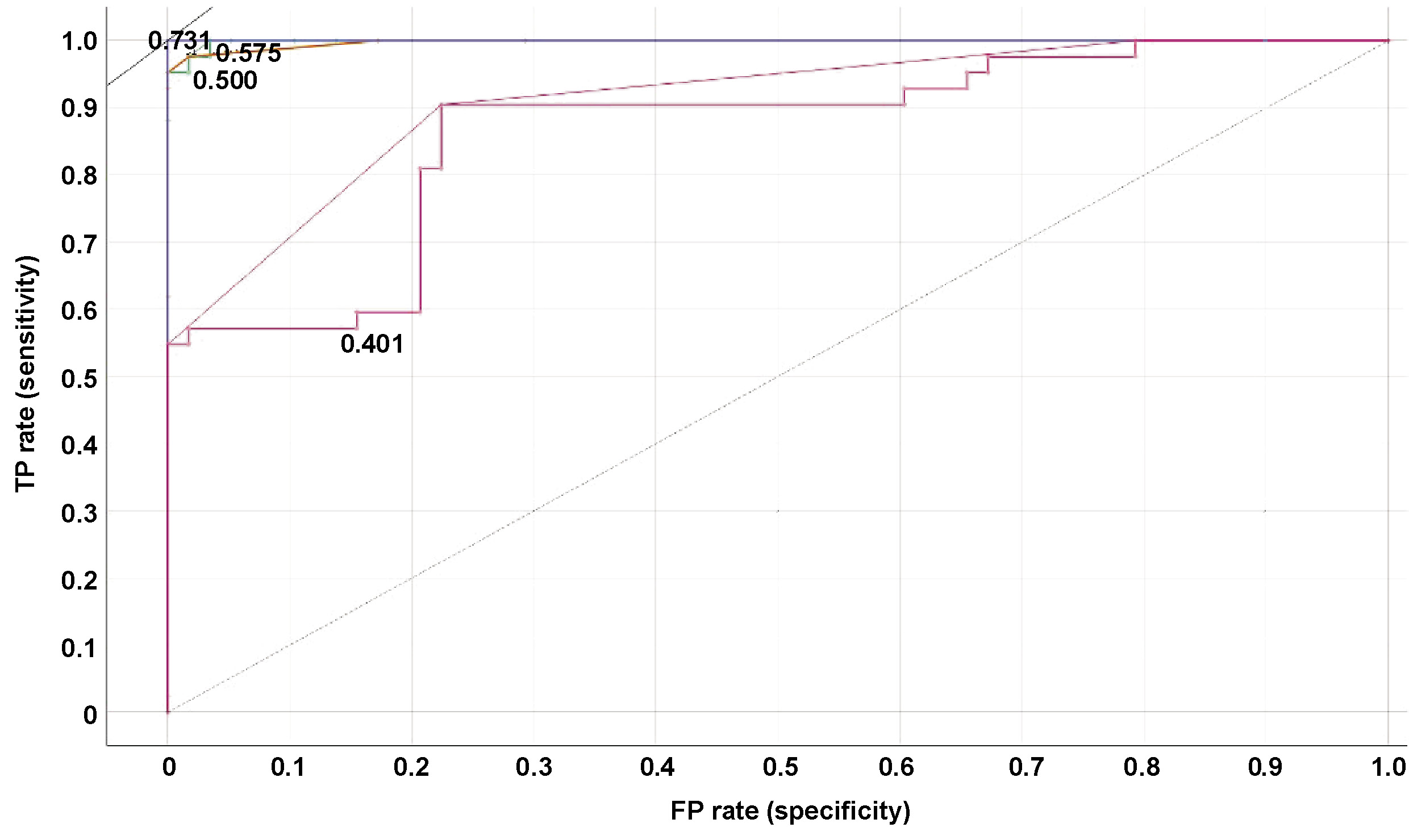
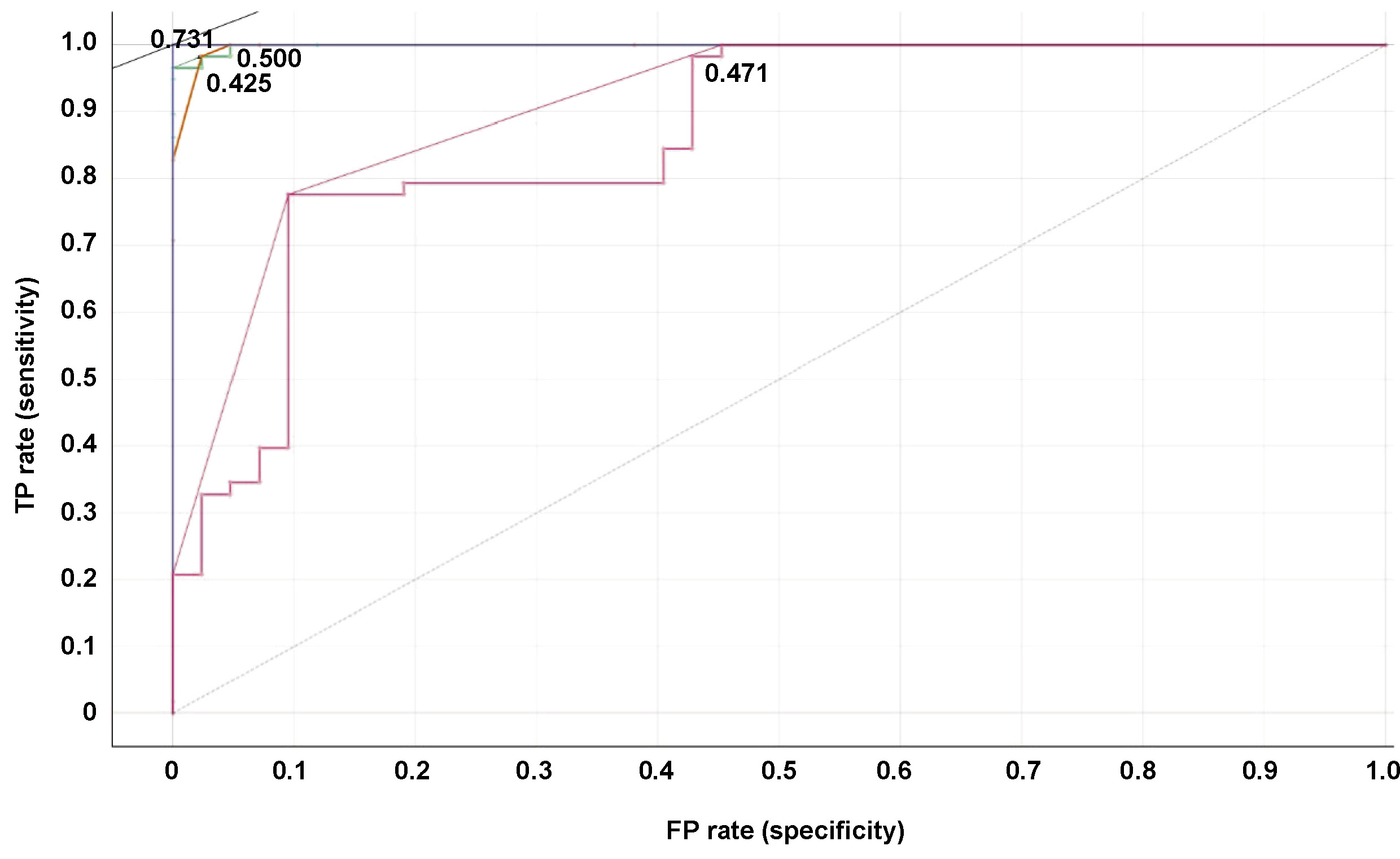
The ratio of correctly identified negative samples to mistakenly recognized positive samples is expressed as TN/FP. The area under the receiver operating characteristic (ROC) curve (AUC) is a useful indicator of a method’s general predictive accuracy (Figure 6, Figure 7).
A significant imbalance was identified between the observed correlations between hub and non-hub genes in diseases (positive cases) and the unobserved correlations (negative cases). In such instances, the effectiveness of a prediction strategy is evaluated using the precision-recall (PR) curve and its area (AUPR). Ideally, the precision parameter for a competent classifier would be set at a value of 1 (high). Precision is defined as 1 when the numerator and denominator are the same, and the FP equals 0 (Equation 3):
In addition to recall, precision, specificity, and accuracy, a confusion matrix is also used to evaluate AUC–ROC curves. The test’s target feature is composed of 2 sets of predictions: hub genes and non-hub genes.
The AUC values obtained for the various models were as follows: 99% for random forest; 99% for decision tree; 100% for AdaBoost; and 86% for neural network (Table 1).
The evaluation of the predicted results using the confusion matrix for the random forest model produced the classification outcomes for the hub gene (Table 2). Similarly, the confusion matrix results for the decision tree model generated the classification results for the predicted hub gene (Table 3).
The assessment of the AdaBoost model using the confusion matrix showed a TP result of 42 for hub genes and a TN count of 58 for non-hub genes (Table 4).
Finally, the evaluation of the neural network model using the confusion matrix yielded a TP value of 26 for hub genes and a TN value of 74 for non-hub genes (Table 5).
Discussion
Periodontitis and oral cancer are diseases with distinct etiologies and pathologies. The identification of similar gene signatures in periodontitis and oral cancer can offer useful insights into the shared mechanisms underlying these diseases.41 Both conditions are associated with persistent inflammation. It has been widely established that oral squamous cell carcinoma (OSCC) is the most prevalent cancer associated with oral bacterial infections. To date, various investigations have revealed the positive effects of Porphyromonas gingivalis on the initiation and development of OSCC.24 Studies have identified several common inflammatory signaling pathways, including the NF-κB route and the mitogen-activated protein kinase (MAPK) pathway. NF-κB regulates gene expression in inflammation, cell survival and the immune response. The activation of the NF-κB pathway has been observed in both periodontitis and oral cancer, contributing to disease development. The MAPK pathway is vital in cell proliferation, differentiation and survival.24, 30, 42, 43, 44 Its dysregulation has been implicated in both illnesses, causing cellular transformation and tissue damage. Understanding the interactome is crucial for the study of hub genes, as they are highly connected within the interactome network. The identification and characterization of these genes can reveal their functions and potential regulatory mechanisms under various biological conditions. Additionally, the definition of the interactome may vary depending on the specific issue or biological system under investigation.
Epigenetic modifications, such as DNA methylation and histone modifications,5, 45, 46, 47 play a significant role in the regulation of gene expression. These changes have linked periodontitis with oral cancer, suggesting the presence of shared underlying processes. Both disorders have been linked to the hypermethylation of tumor suppressor genes, such as p16INK4a (CDKN2A), which renders these genes inactive and contributes to the progression of the disease. The deregulation of gene expression in periodontitis and oral cancer has also been linked to histone changes, such as histone acetylation and methylation.
Both periodontitis and oral cancer induce crucial processes involving the immune system and extracellular matrix remodeling. Common gene profiles linked to immune cell infiltration, cytokine generation and matrix remodeling have been identified in various disorders. Interleukin-6 (IL-6), interleukin-8 (IL-8), and matrix metalloproteinases (MMP-7, -8 and -9), for example, are elevated in both periodontitis and oral cancer, which contributes to tissue invasion and destruction. The dysregulated expression of these genes suggests the presence of shared pathways between immune response dysregulation and remodeling of the extracellular matrix. Cytokines, such as IL-6, have been observed to promote the development of tumors by increasing intracellular reactive oxygen species (ROS) and reactive nitrogen intermediates (RNIs), as well as by altering the epigenetic state of certain genes. Additionally, cytokines promote the development of tumors by activating transcription factors that are associated with tumorigenesis. The production of chemokines is then induced by activated transcription factors, leading to ongoing tumor inflammation.
Both periodontitis and oral cancer frequently exhibit changes in tumor suppressor genes and oncogenes. Both disorders are typically characterized by mutations or inactivation of the well-known tumor suppressor gene TP53, which promotes uncontrolled cell proliferation and genomic instability. Similar changes have been identified in oral cancer and periodontitis in other tumor suppressor genes, including CDKN2A.48 On the other hand, oncogenes such as EGFR and KRAS are frequently dysregulated in oral cancer and have also been linked to periodontitis, indicating the existence of overlapping pathways that promote cell proliferation and survival. A network-based analysis has identified several hub genes that are critical for the initiation and propagation of oral cancer. Within PPI networks, these hub genes frequently display significant levels of connection, indicating their crucial role in regulating cellular processes.
In the present study, interactome hub genes associated with periodontitis and oral cancer were identified using datasets from the GEO database. Initially, 20 cases of periodontitis and oral cancer-specific DEGs were found using a variety of machine-learning approaches. The classification of diagnostic models was developed using random forest, decision tree, AdaBoost, and neural networks. According to the ROC curve, the AUC for random forest was 0.999, for the decision tree was 0.998, for AdaBoost was 1.000, and the neural network model had an AUC of 0.865. The AdaBoost model, followed by random forest and decision tree, exhibited the highest level of accuracy (1.000). The findings indicate that the AdaBoost, random forest and decision tree models have a high diagnostic value and have the potential to facilitate the early detection of periodontitis and oral cancer.49, 50, 51, 52, 53
The cell cycle mitotic pathway, EGFR signaling pathway, electron transport chain signaling pathway, aurora kinase B signaling pathway, and TNF-α signaling via the NF-κB signaling pathway were all found to have enriched DEGs after the identification of the hub genes. This finding suggests that immune response-related pathways likely play a substantial role in the development of periodontitis and oral cancer.
Network analysis techniques, including gene co-expression and functional interaction networks, have been used to identify interactome hub genes in oral cancer and periodontitis. These methods integrate various data types, such as gene expression patterns and functional annotations, to identify highly connected genes and their associated functional relationships. A total of 250 genes associated with periodontitis and 250 genes related to oral cancer were identified among the 500 DEGs. The top 5 hub genes (RSPO4, CDHR2, DDAH2, HLA-J, and IRF3) were identified as priority areas for investigation, with a focus on their relationship to oral cancer and periodontal disease.
The gene RSPO4,45 an activator of canonical Wnt signaling, has been linked to stage III–IV, grade C periodontitis in several European populations, which raises the possibility that this gene plays a role in the development of severe, rapid types of periodontitis. Additionally, RSPO4 regulates interferon-alpha signaling, extracellular matrix interactions, and the mucin barrier.27
A unique biomarker for cancer, CDH2, can be used to study transendothelial migration and inadequate differentiation. Recent studies suggest that N-cadherin plays a significant role in the pathogenesis of hematologic malignancies, including multiple myeloma and leukemia. The expression of the N-cadherin gene (CDH2) is elevated in patients with multiple myeloma who are at high risk for t(4;14)(p16;q32) translocation.54 Furthermore, increasing the expression of CDH2 (rs643555C>T) has been connected to biochemical recurrence of prostate cancer and tumor aggressiveness. CDH2 promotes the epithelial–mesenchymal transition, stemness and metastatic potential of prostate cancer cells by stimulating the ErbB signaling pathway. Additionally, the DDAH2 gene (chromosome 6p21.3) produces an enzyme that regulates the levels of methyl arginine within cells, thereby facilitating the synthesis of nitric oxide (NO). This, in turn, impedes the activity of nitric oxide synthase (NOS) in healthy cells. A candidate for a hypermethylated gene with downregulated protein expression in OSCC, the DDAH2 gene, appears to play a crucial role in the development of cancer.
Due to defects in the gene and a lack of any associated functional activity, HLA-J, otherwise known as the major histocompatibility complex, class I, J (pseudogene),55 has long been acknowledged as a pseudogene. However, a recent study found functionally significant transcriptional activity in breast cancer patients.57 Immunosuppressive proteins HLA-G and HLA-J exhibit a high degree of sequence homology. This provides a starting point for deducing the functional relevance of HLA-J in infection-induced antinociception, particularly in females. According to earlier investigations, IRF3 is functionally implicated in producing cytokines and chemokines in response to the P. gingivalis challenge, which leads to the activation of IRF3. The host response to P. gingivalis activates IRF3, and IRF3 ablation reduces TNF production in response to P. gingivalis.33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49
Earlier studies have shown that IRF3 plays a functional role in driving cytokine and chemokine production in response to P. gingivalis.42 Upon exposure to P. gingivalis, IRF3 becomes activated. As previously established, the host response to P. gingivalis activates IRF3, and IRF356 ablation has been demonstrated to reduce TNF production in response to P. gingivalis.
The primary objective of future research should be to advance our understanding of the complex interactome networks in periodontitis and oral cancer.57, 58, 59 Integrating multi-omics data, including genomics, trainscriptomics and proteomics will enable a more comprehensive perspective on the underlying mechanisms of the diseases. Collaboration is also necessary to create sizable, well-annotated datasets that can be used for reliable forecasts and validations.
Conclusions
The prediction of interactome hub genes in periodontitis and oral cancer using network-based techniques is a promising direction. The development of targeted treatments and the identification of potential biomarkers would be facilitated by the ability to identify these key genes and their relationships. Further investigation and validation are necessary to fully comprehend the complex molecular networks underlying oral cancer and periodontitis. This knowledge will pave the way for individualized therapies and enhance patient outcomes in the future.
Ethics approval and consent to participate
Not applicable.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Use of AI and AI-assisted technologies
The authors used AI-assisted tools (Grammarly, Reverso, ChatGPT, OpenAI, and similar language models) exclusively for language refinement, grammar correction, and enhancing the clarity of writing. No AI tools were used for data analysis, interpretation, statistical procedures, or generation of scientific content. All scientific conclusions were made by the authors.