Abstract
Background. Antimicrobial resistance (AMR) must be predicted to combat antibiotic-resistant illnesses. Based on high-priority AMR genomes, it is possible to track resistance and focus treatment to stop global outbreaks. Large language models (LLMs) are essential for identifying Porhyromonas gingivalis multi-resistant efflux genes to prevent resistance. Antibiotic resistance is a serious problem; however, by studying specific bacterial genomes, we can predict how resistance develops and find better kinds of treatment.
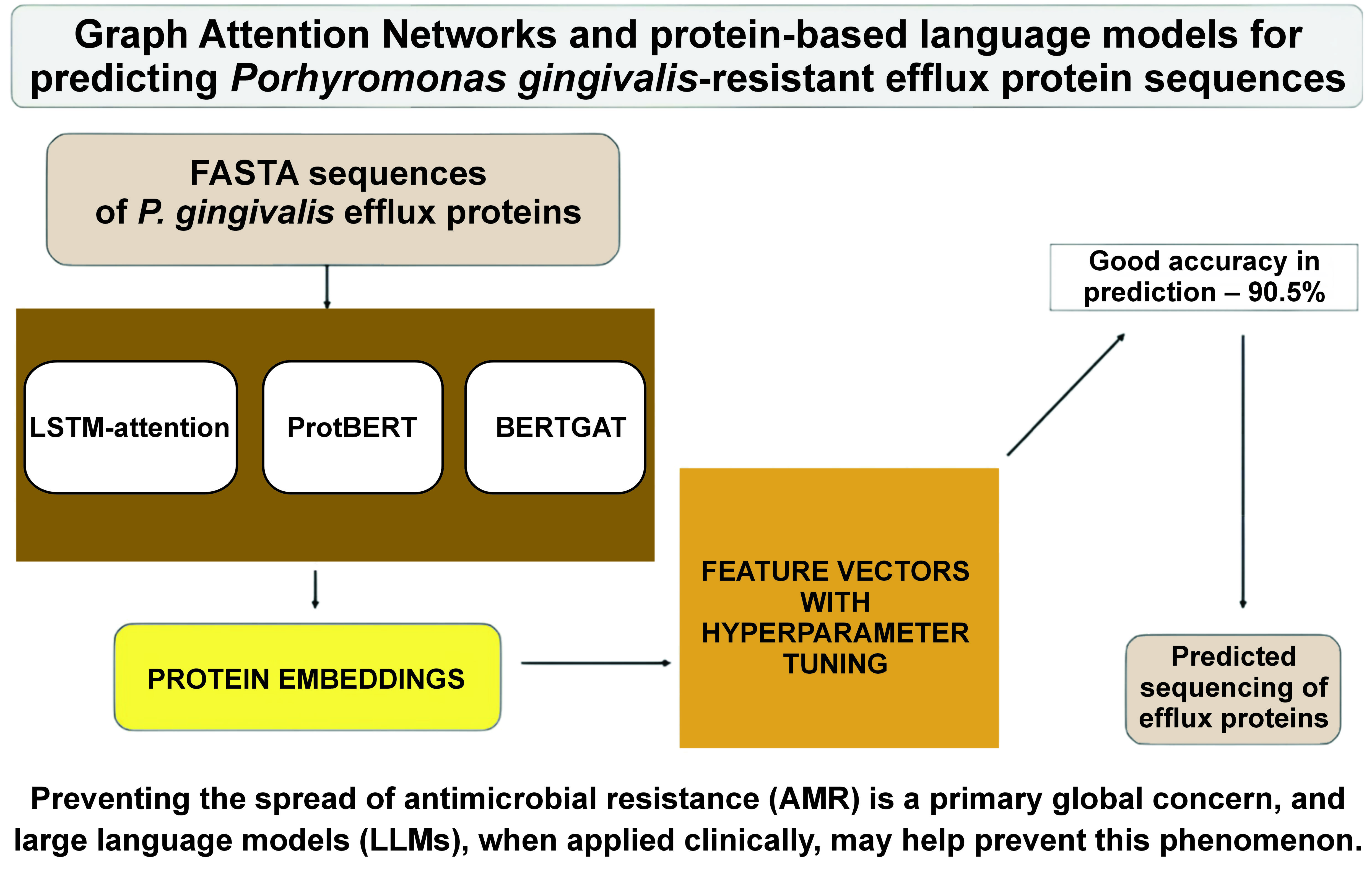
Objectives. This paper explores using advanced models to predict the sequences of proteins that make P. gingivalis resistant to treatment. Understanding this approach could help prevent AMR more effectively.
Material and methods. This research utilized multi-drug-resistant efflux protein sequences from P. gingivalis, identified through UniProt ID A0A0K2J2N6_PORGN, and formatted as FASTA sequences for analysis. These sequences underwent rigorous detection and quality assurance processes to ensure their suitability for computational analysis. The study employed the DeepBIO framework, which integrates LLMs with deep attention networks to process FASTA sequences.
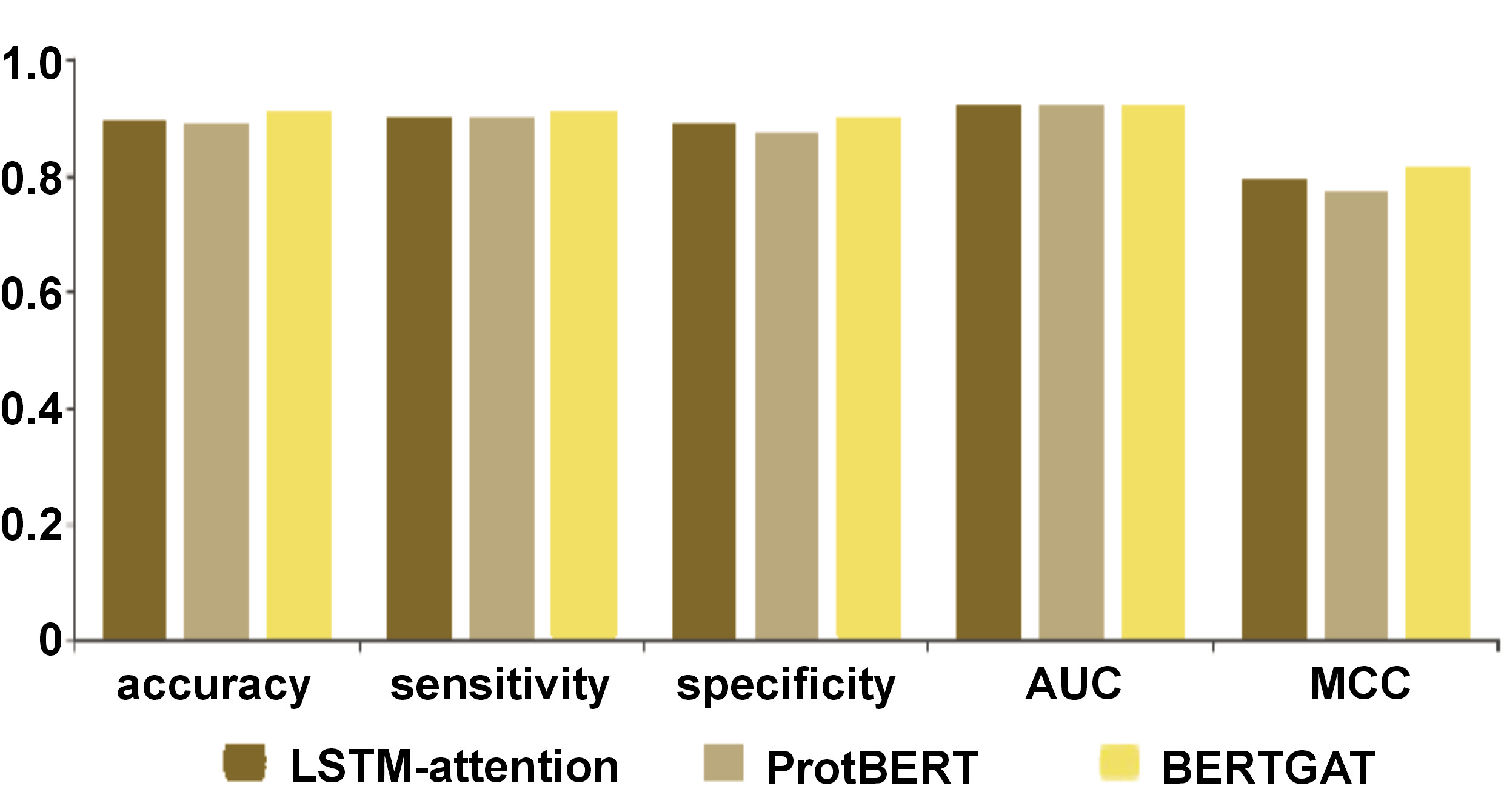
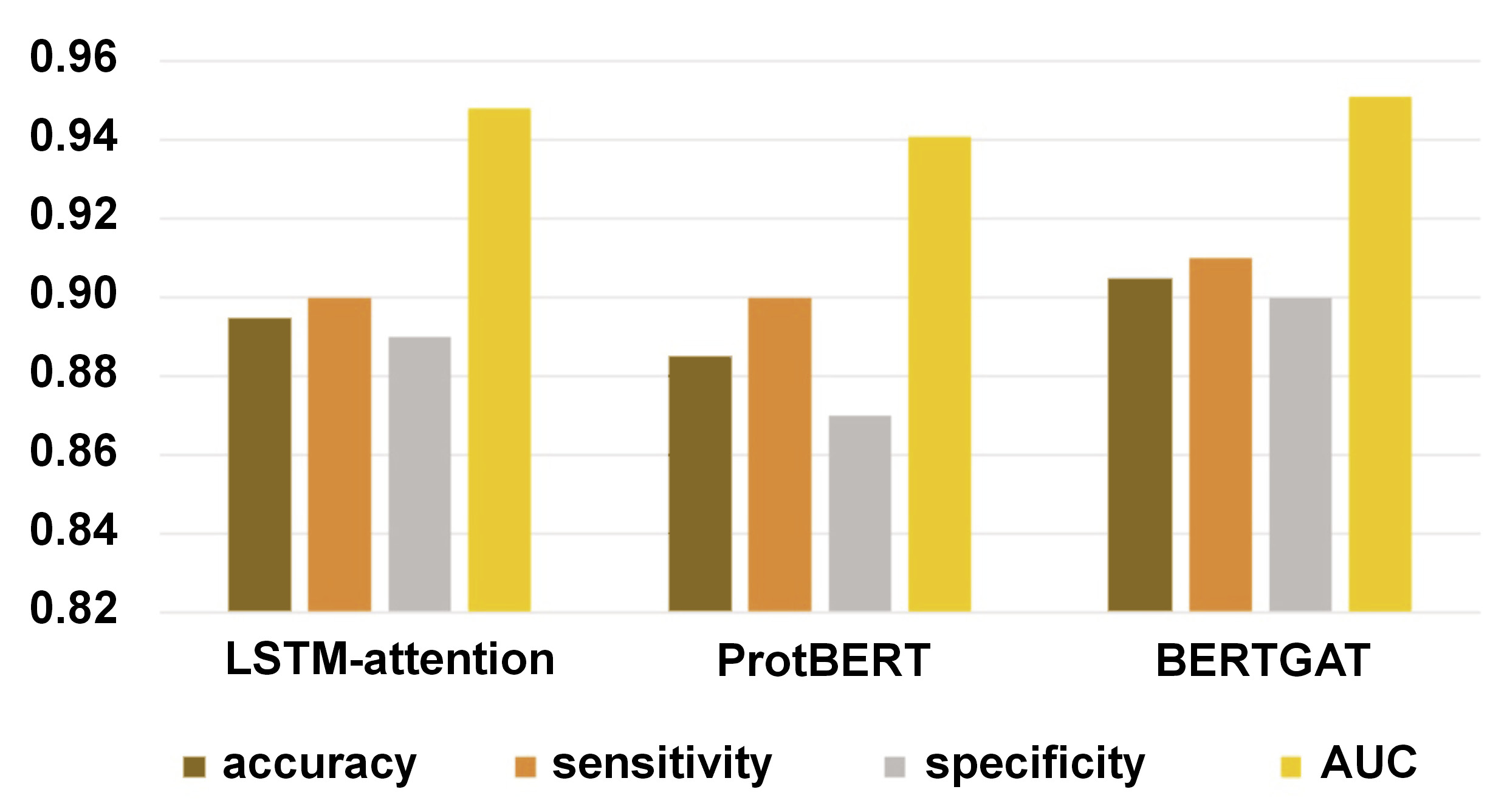
Results. The analysis revealed that the Long Short-Term Memory (LSTM)-attention, ProtBERT and BERTGAT models achieved sensitivity scores of 0.9 across the board, with accuracy rates of 89.5%, 88.5% and 90.5%, respectively. These results highlight the effectiveness of the models in identifying P. gingivalis strains resistant to multiple drugs. Furthermore, the study assessed the specificity of the LSTM-attention, ProtBERT and BERTGAT models, which achieved scores of 0.89, 0.87 and 0.90, respectively. Specificity, or the genuine negative rate, measures the ability of a model to accurately identify non-resistant cases, which is crucial for minimizing false positives in AMR detection.
Conclusions. When utilized clinically, this LLM approach will help prevent AMR, which is a global problem. Understanding this approach may enable researchers to develop more effective treatment strategies that target specific resistant genes, reducing the likelihood of resistance development. Ultimately, this approach could play a pivotal role in preventing AMR on a global scale.
Keywords: periodontitis, antimicrobial resistance, large language models, Porphyromonas gingivalis, efflux protein
Introduction
Antimicrobial resistance (AMR)1, 2 is the ability of microorganisms to resist the effects of antimicrobial drugs, such as antibiotics, antivirals and antiparasitics.3, 4, 5 Combating antibiotic-resistant diseases requires predicting AMR. High-priority AMR genomes can lead surveillance to track resistance and focus treatment in order to prevent global outbreaks.6, 7, 8
Leveraging insights from large language models (LLMs), like ProtBERT or BERTGAT, can be employed to explore the intricate mechanisms governing the interplay between protein sequences, their structural configurations and resultant functions.9, 10 The essence of this paradigm lies in understanding how the linear arrangement of amino acids, akin to the syntax of a sentence, dictates the three-dimensional (3D) structure of a protein, which, in turn, governs its biological functions. By adopting computational language models, traditionally used in natural language processing (NLP), we gain a valuable tool to dissect and decipher the functions of proteins.11, 12, 13 This approach allows researchers to unveil the nuanced relationships between amino acid sequences, the structural motifs they form and the functional roles they play in biological processes. Treating protein sequences as linguistic entities provides a powerful framework for unraveling the language of life encoded in these fundamental biological molecules.14
The attention-based Long Short-Term Memory (LSTM-attention) network is a method that analyzes big datasets and looks for patterns that point to AMR, using state-of-the-art algorithms.15, 16, 17, 18, 19, 20, 21, 22 Co-AMPpred is one instance of a machine learning method for AMR prediction.23, 24 This tool distinguishes between antimicrobial peptides (AMPs) and non-AMPs by combining physicochemical characteristics and composition-based sequences through machine learning techniques.
An important global health concern is periodontitis, an immune-inflammatory infectious disease, mostly caused by Porphyromonas gingivalis.25, 26 The bacterium exhibits a variety of omics and phylogeny information, making it a significant factor in severe periodontitis. Treatment for P. gingivalis is becoming more difficult due to its growing resistance to antibiotics, which highlights the need for a deeper comprehension of its resistance mechanisms. In particular, the resistance-nodulation-division (RND) family of efflux pumps is a major contributor to the AMR of P. gingivalis. These pumps, including proteins such as AcrA, AcrB and TolC,27, 28, 29, 30 block the entry of antimicrobial drugs into the bacterial cell, contributing to multi-drug resistance (MDR).
Porphyromonas gingivalis-produced gingipains and virulence factors31, 32 add to the complexity of the situation. Due to gingipains, P. gingivalis can elude the host immune system, which contributes to AMR. The integrated protein–protein interaction network (PPIN), which includes virulence regulators and efflux pump proteins, was subjected to topological and functional analysis; this analysis identified genes crucial for understanding the relationships across cellular systems in P. gingivalis.31 The bifunctional NAD(P)H-hydrate repair enzyme A0A212GBI3_PORGN is one of the most prevalent resistant efflux proteins.33, 34, 35, 36, 37 It is essential for the bifunctional enzyme that it catalyzes the dehydration of the S-form of NAD(P)HX38 at the expense of ADP, which is converted to AMP, as well as the epimerization of the S- and R-forms of NAD(P)HX.
Identifying P. gingivalis multi-resistant efflux genes with the use of LLMs is crucial for preventing resistance. The present study aimed to analyze and explore Graph Attention Networks (GATs) and protein-based language models for predicting P. gingivalis resistant efflux protein sequences.
Methods
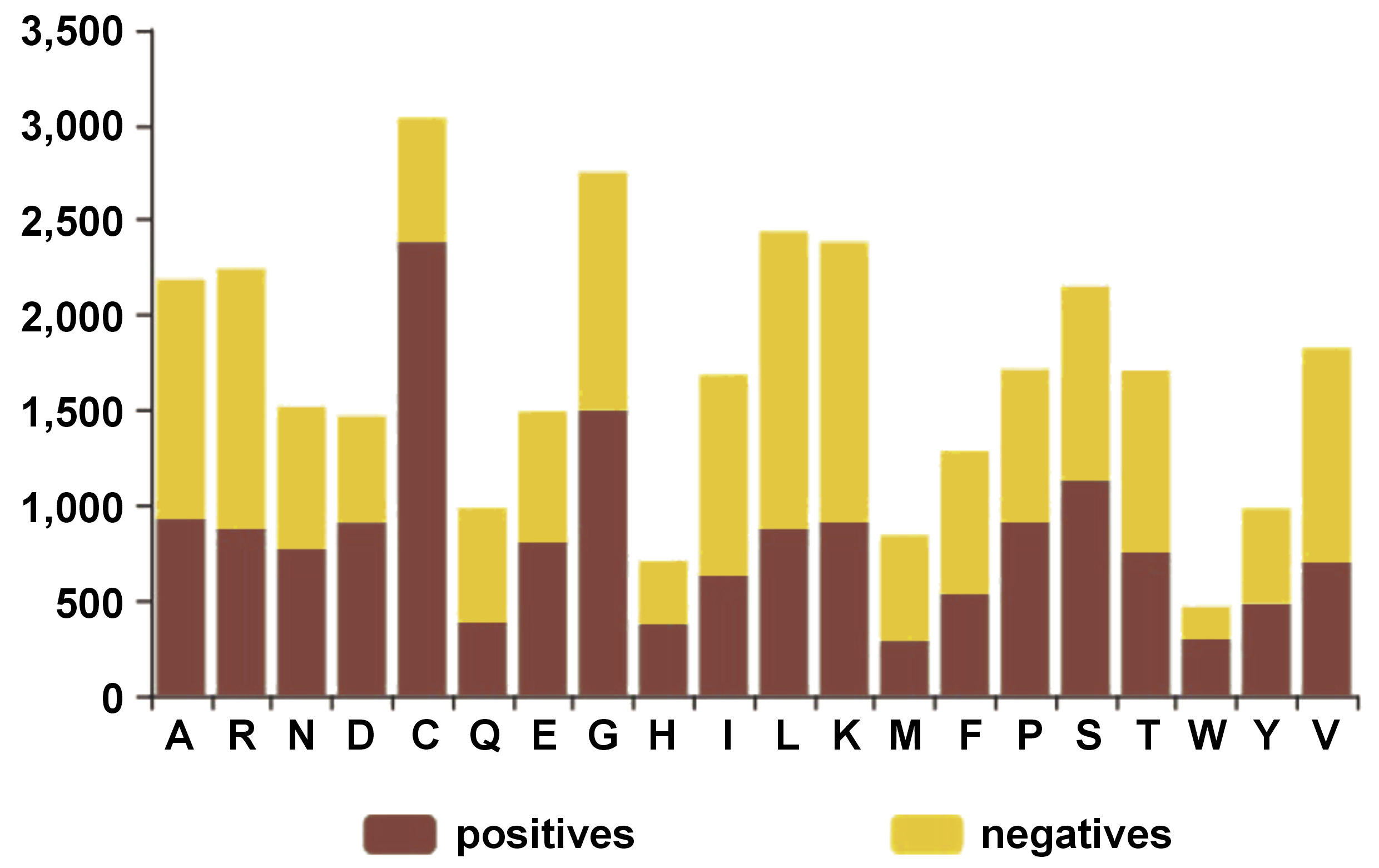
Using UniProt,39 the following sequences of multi-drug resistant proteins of P. gingivalis were downloaded: A0A0K2J2N6_PORGN; A0A212GBI3_PORGN; A0A2D2N4E3_PORGN; A0A0E2LNT1_PORGN; A0A829KLL9_PORGN; U2K1P7_PORGN; Q7MXT9_PORGI; A0A1R4DUJ6_PORGN; and A0A212FQN2_PORGN. The identified FASTA sequences underwent a thorough quality check to ensure that there were no biases during their entry. Additionally, the sequences were formatted according to the prescribed format based on the DeepBIO tool for LLMs and deep attention networks.40
DeepBIO
Academics can construct a deep learning architecture to address any biological problem with the help of DeepBIO, a one-stop web service. In addition to visualizing biological sequencing data, DeepBIO compares and enhances deep learning models. It offers base functional annotation tasks, with in-depth interpretations and graphical visualizations, and conservation motif analysis to confirm site dependability, and well-trained deep learning architectures for more than 20 tasks. The sequence-based datasets were divided into the training and test sets using DeepBio. We randomly divided each dataset into 1,000 training and 200 testing sets to optimize hyperparameters and analyze performance.
BERTGAT
BERTGAT41 is a neural network model that combines the pre-trained language model Bidirectional Encoder Representations from Transformers (BERT) with GAT.16, 42 BERT extracts text features,41 while GAT learns the sentence–word relationships.26, 43, 44 Transformer-based language models are preferred over recurrent neural networks (RNNs). Pre-trained BERT representations are fine-tuned to generate state-of-the-art models for wide-ranging text-to-structured query language (SQL) workloads with one extra output layer.
ProtBERT
The provided search results do not contain specific information about the full code architecture of ProtBERT and its detailed steps. However, based on the available information, it was possible to provide a general outline of the architecture and the steps involved in using ProtBERT for protein sequence prediction.41
ProtBERT architecture and steps for protein sequence prediction
Pre-training
ProtBERT is pre-trained on a large dataset of protein sequences, representing the entire known protein space, using a masked language modeling task combined with a novel Gene Ontology (GO) annotation prediction task. The architecture of ProtBERT consists of local and global representations, allowing the end-to-end processing of protein sequences and GO annotations.
Fine-tuning
After pre-training, the ProtBERT model is fine-tuned on specific protein-related tasks, such as protein sequence classification or function prediction. Fine-tuning involves initializing the model from the pre-trained state, freezing some layers, training additional, fully connected layers, and then unfreezing all layers for further training.
Model evaluation
The fine-tuned ProtBERT model is evaluated on diverse benchmarks covering various protein properties to assess its performance. The ProtBERT model is built on Keras/TensorFlow and is available through the Hugging Face model hub. The code for using ProtBERT involves loading the pre-trained model, fine-tuning it on specific protein-related tasks, and utilizing it for protein sequence prediction and analysis.
LSTM-attention model
LSTM15, 17 and attention mechanisms are combined in LSTM-attention, a deep learning architecture, to enhance sequence prediction task performance. The following steps are needed to put the LSTM-attention model into practice:
1. Data Preparation: The first stage is to prepare the input data for the model. This could entail activities like feature extraction, encoding and tokenization.
2. Model Architecture: An LSTM layer and an attention layer form the LSTM-attention model. After processing the input sequence, the LSTM layer creates a series of hidden states. The more pertinent states are given more weight when the attention layer computes a weighted sum of the hidden states.
3. Training: The model is trained using the appropriate loss function and optimization technique with the prepared data. The parameters of the model are adjusted during training to minimize the loss function.
4. Evaluation: After training, the performance of the model is assessed on an independent test set. This entails calculating metrics like the F1 score, recall, accuracy, and precision.
5. Prediction: The model can forecast new sequences after evaluation. The trained model receives the input sequence and the learned weights generate the output.
6. Fine-tuning: The model can be further adjusted on particular tasks or datasets to boost performance. This involves changing the hyperparameters or architecture of the model to fit a given task better (Table 1).
Results
LSTM-attention, ProtBERT and BERTGAT were used to find the hidden features and weights in the FASTA protein sequences; then, backpropagation algorithms with ADAM optimizer and 50 iterations fine-tuned the model.
LSTM-attention, ProtBERT and BERTGAT had sensitivity of 0.90, 0.90 and 0.91, respectively (TP / (TP + FN); TP – true positive, FN – false negative). Specificity, or the true negative rate, is the proportion of actual negatives correctly predicted as negatives. The specificity of LSTM-attention, ProtBERT and BERTGAT was 0.89, 0.87 and 0.90, respectively (TN / (TN + FP); TN – true negative, FP – false positive).
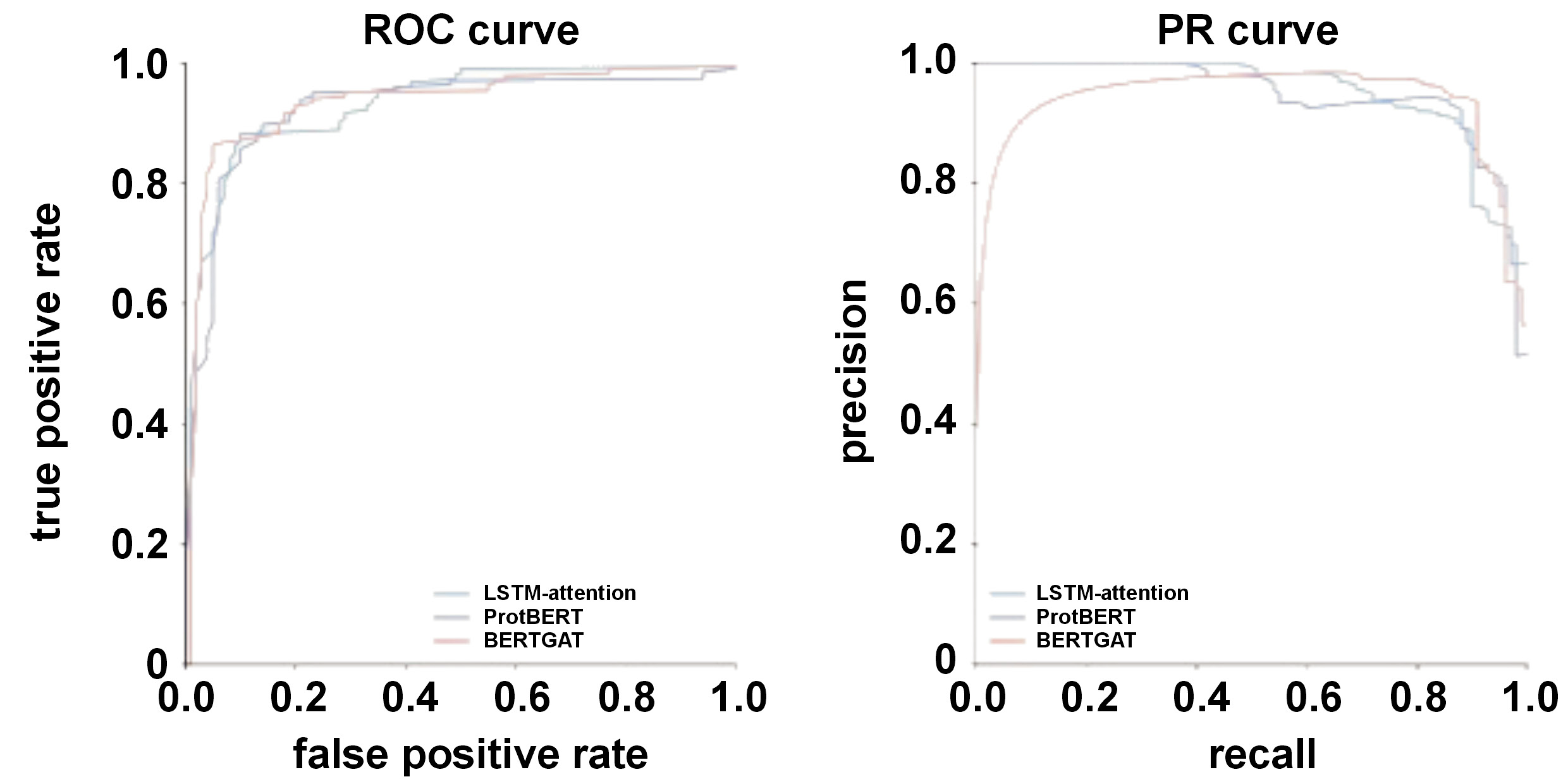
ROC curve
The receiver-operating characteristic (ROC) curve shows the trade-off between the true positive rate (sensitivity) and false positive rate (1-specificity) of the model over the categorization thresholds. Regarding LSTM-attention, ProtBERT and BERTGAT, high true positive rates are shown by the ROC curve in the upper left corner of the plot.
PR curve
The trade-off between recall and precision for binary classifiers with different probability thresholds is depicted by the precision–recall (PR) curve. While precision is the fraction of positive predictions, recall is the percentage of accurately expected positives. This model’s performance with uneven classes is made public. The area under the PR curve (AUC-PR) is a widely used metric to summarize the classifier performance. Higher AUC-PR values for LSTM-attention, ProtBERT and BERTGAT denote improved model performance.
An epoch plot is a graph showing the accuracy and loss of a machine learning model over training. It is an effective diagnostic tool for overfitting and other model issues. The number of epochs or iterations the model has been trained on is shown by the X-axis in an epoch plot. The accuracy or loss of the model is plotted on the Y-axis. The loss indicates how effectively the model predicts the proper output for a given input. Accuracy gauges whether the predictions of the model are accurate.
UpSet plot
The frequency of common items between groups can be ascertained by comparing the intersection diameters. While smaller crossings imply less overlap, larger intersections show more overlap between groups. In a vertical UpSet plot, rows represent intersections and matrix columns represent sets. Each row has filled intersection cells that show how the rows are related to each other.
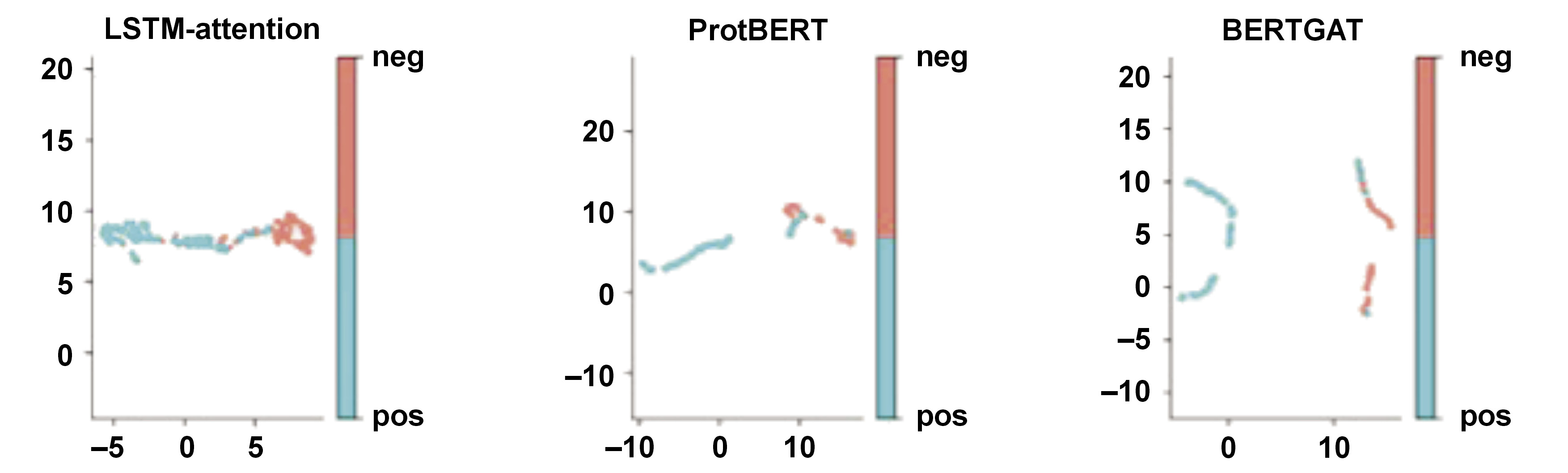
Uniform Manifold Approximation and Projection (UMAP) creates a weighted graph from high-dimensional data to show clustering patterns, with the edge strength reflecting how ‘close’ the points are. Projecting this graph lowers its dimension. This data shows algorithm clustering. UMAP is a non-linear dimension reduction method for embedding high-dimensional data in low-dimensional space. It assumes that high-dimensional data points should be close to low-dimensional space.
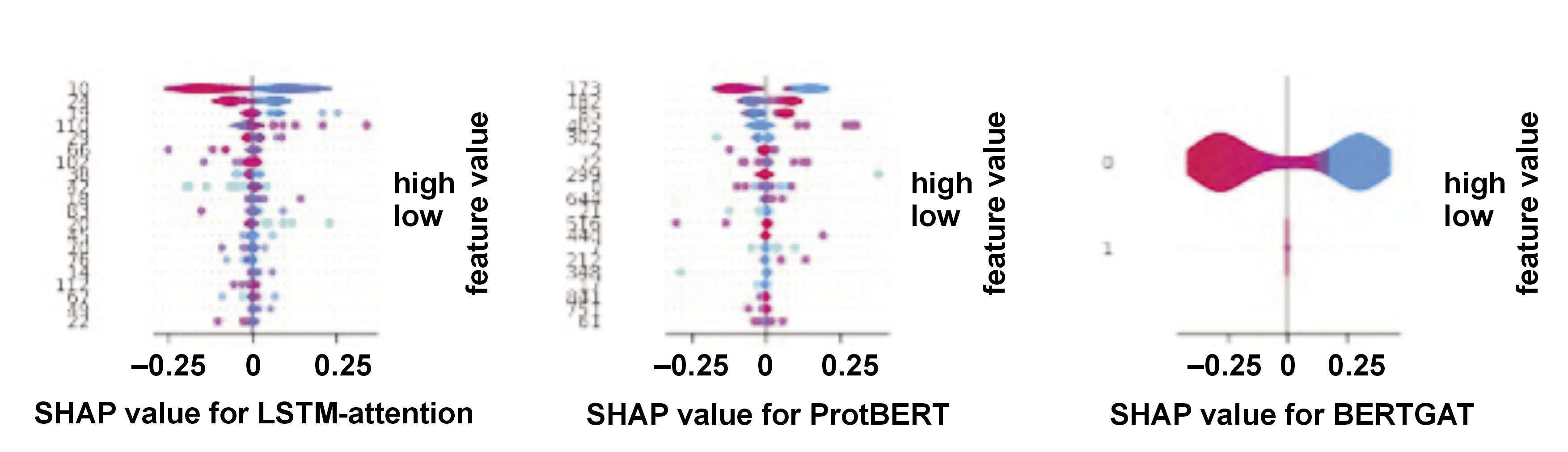
SHAP values
The predictive value of each feature is quantified in a machine learning model. All possible feature combinations are considered, along with the relative contributions of each feature to the prediction when coupled with a subset of features, to compute the value. When a feature enhances the prediction, the Shapley Additive Explanations (SHAP) red value is positive. A feature with a negative SHAP blue value is less predictive.
Discussion
Antimicrobial drug-resistant periodontal bacteria45, 46, 47 are characterized by efflux pumps – proteins that remove antimicrobial medications from the cell, thus preventing the drugs from killing the bacteria. Bacteria can also adapt their outer membrane to block antimicrobial medications or change the target site of the drug to lessen its efficacy.36, 48 These pathways and others cause antibiotic resistance in periodontitis patients. Whole-genome sequencing can detect AMR genes34, 35, 49, 50 and mutations, assessing the resistance potential. Large genomic, phenotypic and clinical datasets can be used to train machine learning algorithms to predict resistance and discover the key AMR genes. Prolonged illness, more expensive second-line therapies and missed productivity can strain healthcare systems and national economies. Predicting AMR in globally prevailing periodontal infections, especially for the keystone pathogens like P. gingivalis, is important for preventing resistance from spreading across continents. Antimicrobial resistance is a growing concern in the field of periodontitis research. It refers to the ability of microorganisms, such as bacteria, to resist the effects of antimicrobial drugs.1, 51
Large language models have revolutionized various fields, including protein sequence prediction. In this study, models such as LSTM-attention, ProtBERT and BERTGAT demonstrated high predictive performance, with accuracy rates reaching up to 90.5% (Table 2, Figure 1, Figure 2, Figure 3, Figure 4, Figure 5). Large language models have also shown strong results in broader protein-related tasks, such as structure prediction and protein design, in previous studies.
The observed performance differences between LSTM-attention, ProtBERT and BERTGAT, with accuracy rates of 89.5%, 88.5% and 90.5%, respectively, deepen the interpretation of the results in the context of model architecture implications.52, 53 LSTM-attention utilizes long short-term memory units and attention mechanisms, while ProtBERT incorporates a transformer-based architecture, specifically designed for protein sequence data, and BERTGAT incorporates graph attention mechanisms. The higher accuracy of BERTGAT suggests that its increased model complexity and ability to capture graph structures in the data have contributed to improved performance. Data representation is another important factor to consider. The comparable accuracy of LSTM-attention and ProtBERT suggests that their respective data representations are effective for a given task. Biological relevance is a critical consideration when evaluating model performance. Protein sequence analysis is inherently tied to biology, and it is important to assess how well the models align with biological knowledge.50, 52, 54 While all 3 models demonstrated high accuracy, it is necessary to delve deeper into the interpretation to understand if the superior accuracy of BERTGAT is biologically relevant or if other factors drive it. Overall, the observed performance differences between LSTM-attention, ProtBERT and BERTGAT highlight the impact of model complexity, data representation and biological relevance. Further analysis and interpretation are required to uncover the specific advantages of each architecture and their implications in the context of protein sequence analysis.
Previous state-of-the-art models, like ProteinBERT,55, 56, 57 a universal deep learning model for protein sequences, leveraging the transformer architecture,58, 59, 60 are commonly used in NLP tasks. In addition to language models, various machine learning methods and algorithms are used in protein sequence prediction, such as graph neural networks and deep learning-based algorithms like BERTGAT and LSTM-attention.15, 16, 17 ProtBERT is a transformer-based language model trained on a large corpus of protein sequences to learn representations that capture important structural and functional information.24 This study compared LLMs vs. GAT-based algorithms57, 61, 62 in predicting AMR sequencing, and the performance of the model was shown using the SHAP,63, 64, 65 UMAP and UpSet plot analysis (Figure 6, Figure 7, Figure 8), similar to previous studies for performance.
Targeting P. gingivalis efflux proteins is important for novel antibiotic drug design. These prediction models could point to resistance mutation sequences and prevent the development of AMR in periodontitis patients.66, 67
This study compared the performance of LLMs and GAT-based algorithms in predicting AMR sequencing. The model’s performance was evaluated using the SHAP, UMAP and UpSet plot analysis, previously employed to assess the performance of similar prediction models. The study also highlighted the significance of targeting P. gingivalis efflux proteins to design novel antibiotic drugs. However, it is important to acknowledge that the current study has limitations. One major limitation is the small sample size and the lack of the external validation of the independent datasets used in the study.52 Future research should address this limitation by including larger sample sizes to ensure the reliability and generalizability of the prediction model. Furthermore, further investigations are needed to validate the model’s performance in diverse datasets and to explore its applicability for other oral microbes.
Conclusions
Preventing the spread of antimicrobial resistance (AMR) is a primary global concern, and large language models (LLMs), when applied clinically, may help prevent this phenomenon.
Ethics approval and consent to participate
Not applicable.
Data availability
The datasets supporting the findings of the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Use of AI and AI-assisted technologies
Not applicable.