Abstract
Background. Dental biofilms, primarily driven by Streptococcus mutans, are the main etiological agents of caries and restorative failure.
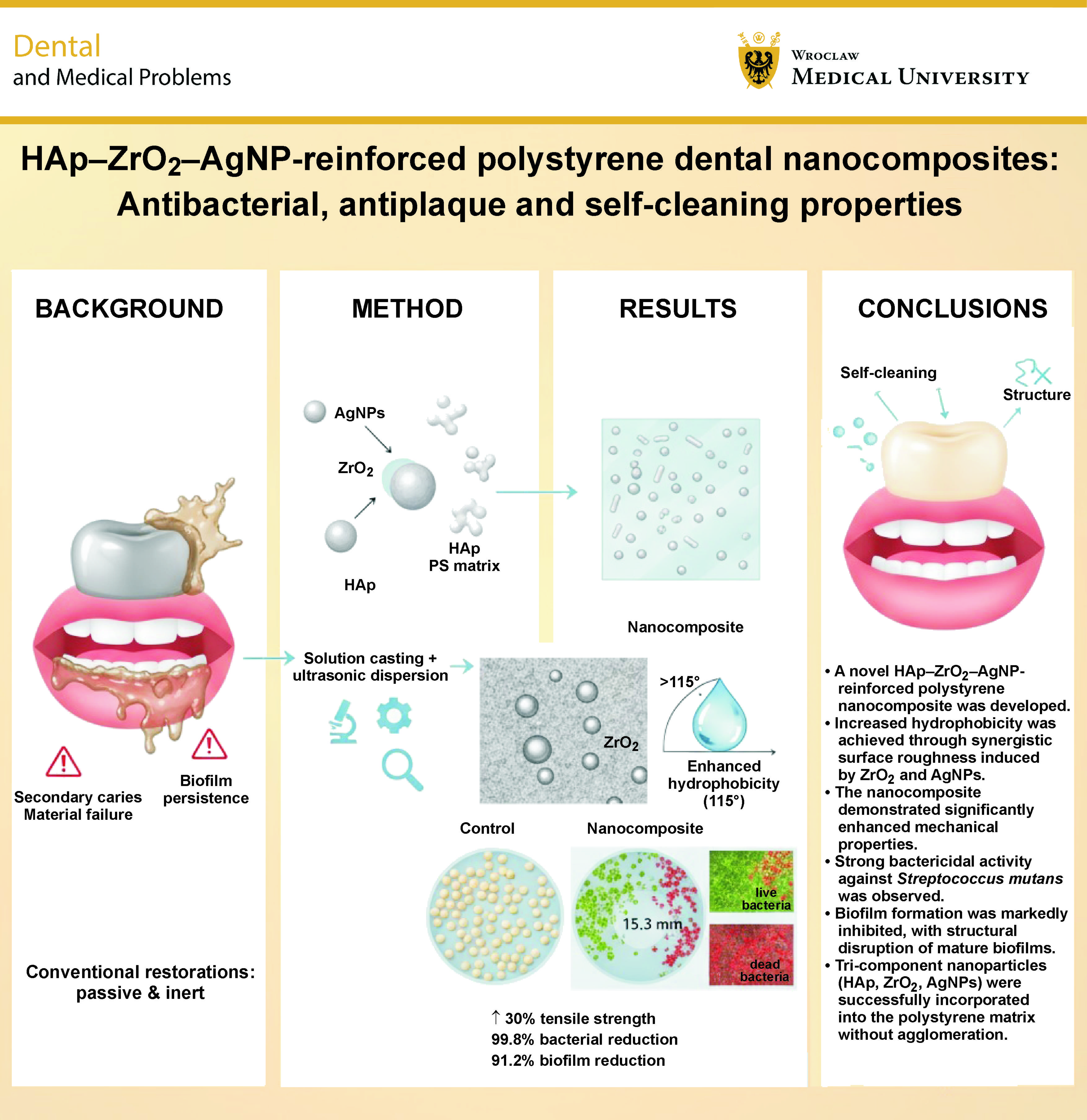
Objectives. The aim of the study was to design and synthesize a novel polystyrene (PS)-based nanocomposite reinforced with hydroxyapatite (HAp), zirconia (ZrO2) and silver nanoparticles (AgNPs).
Material and methods. Hydroxyapatite, ZrO2 and AgNPs were synthesized via wet precipitation, sol–gel and citrate reduction methods, respectively. The nanoparticles (5 wt% total loading) were uniformly dispersed in a toluene-based PS matrix using ultrasonic-assisted solution casting. The resulting films were characterized in terms of morphology (scanning electron microscopy (SEM), transmission electron microscopy (TEM), atomic force microscopy (AFM)), composition (energy-dispersive X-ray spectroscopy (EDX)), thermal stability (thermogravimetric analysis (TGA)), mechanical properties (tensile strength, Young’s modulus, Vickers hardness), surface wettability (water contact angle (WCA)), antibacterial efficacy (zone of inhibition (ZOI), minimum inhibitory concentration/minimum bactericidal concentration (MIC/MBC), colony-forming unit (CFU) assay), and antibiofilm activity (crystal violet staining, confocal laser scanning microscopy (CLSM)). Statistical analysis was performed using one-way analysis of variance (ANOVA) and Tukey’s honestly significant difference (HSD) test (p < 0.05).
Results. The nanocomposite exhibited a 30% increase in tensile strength (32.5 MPa vs. 25 MPa for pure PS) and a 25% rise in elastic modulus (1.5 GPa vs. 1.2 GPa). The water contact angle increased from 85° ±2° (pure PS) to 115° ±3° (nanocomposite), confirming significantly enhanced hydrophobicity. The composite demonstrated strong antibacterial activity, with a ZOI of 15.3 mm, MIC of 6.25 mg/mL, MBC of 12.5 mg/mL, and 99.8% bacterial reduction within 24 h. Antibiofilm performance was also notable, showing a 91.2% reduction in biofilm biomass after 72 h and 74.8% decrease in biofilm thickness (from 32.5 µm to 8.2 µm). Confocal laser scanning microscopy revealed a marked shift in the live/dead cell ratio (from 4.7:1 to 0.6:1), confirming bactericidal rather than bacteriostatic action. All nanoparticles were homogeneously distributed without agglomeration, as confirmed by SEM/EDX and TEM.
Conclusions. The HAp–ZrO2–AgNP-reinforced PS nanocomposite integrates enhanced mechanical properties, self-cleaning surface characteristics, and potent, long-lasting antibiofilm activity against S. mutans. While further biocompatibility and clinical translation studies are warranted, this material represents a significant leap toward durable, infection-resistant dental applications.
Keywords: antibacterial, zirconia, hydroxyapatite, silver nanoparticles, dental biofilms
Introduction
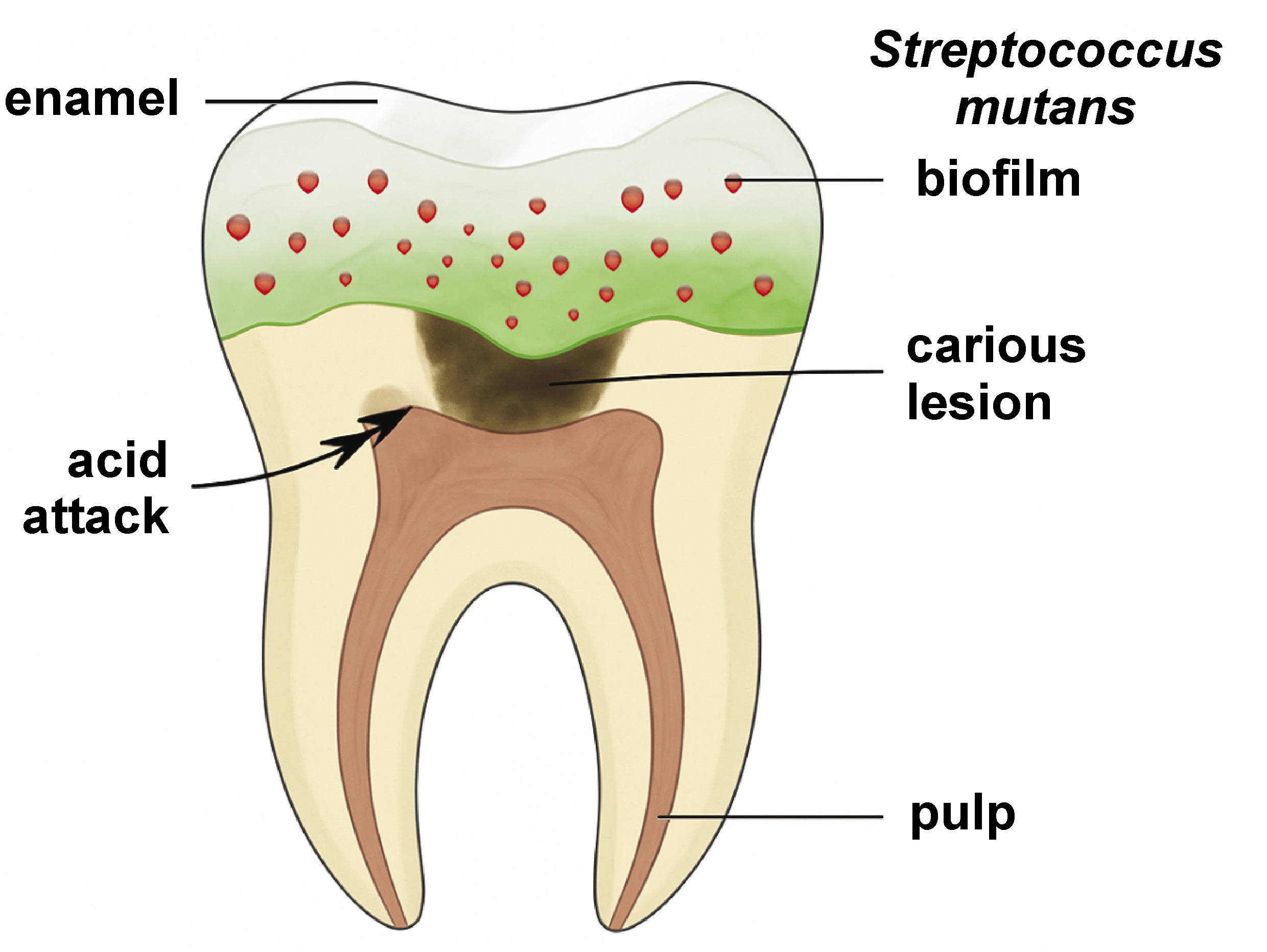
Dental caries and periodontal diseases remain among the most prevalent chronic conditions worldwide, affecting nearly 3.5 billion people according to the World Health Organization (WHO).1, 2, 3 The etiology of dental caries is primarily attributed to dental plaque, a complex biofilm formed by microbial communities adhering to tooth surfaces. The pathogenesis of caries involves acid-producing bacteria (e.g., Streptococcus mutans, Lactobacillus spp.), which metabolize dietary sugars and induce demineralization of tooth enamel (Figure 1).4, 5, 6, 7, 8 Periodontal diseases, on the other hand, are mainly associated with gram-negative anaerobes (e.g., Porphyromonas gingivalis, Fusobacterium nucleatum), which trigger inflammatory responses and tissue destruction.9, 10
Conventional preventive strategies, such as mechanical plaque removal (toothbrushing and flossing) and the use of antimicrobial agents (fluoride, chlorhexidine), present several limitations. Mechanical methods often fail to reach inaccessible areas, while prolonged use of antimicrobial agents may disrupt the balance of the oral microbiota and promote resistance.6, 7 Additionally, dental restorative materials (e.g., composites, resins) lack inherent antibacterial properties, making them susceptible to secondary caries and biofilm accumulation.11, 12, 13, 14, 15
Recent studies have explored various approaches to enhance the antibacterial properties of dental materials. For instance, modification of glass ionomer cements (GICs) with polyamidoamine (PAMAM) and bioactive glass (BAG) has been shown to significantly reduce S. mutans colonization and promote ion release, indicating improved antibacterial efficacy.16 Similarly, natural compounds, such as Zataria multiflora extract, have demonstrated inhibitory effects against oral microorganisms and plaque formation, suggesting potential alternatives to traditional antimicrobials.17 Nanochitosan-modified GICs have also been investigated for their antibacterial and preventive effects on initial enamel caries lesions, showing promising results in terms of microshear bond strength and antibacterial activity.18 Furthermore, natural agents such as propolis and hesperidin have been compared with synthetic agents (e.g., silver diamine fluoride) in deep carious dentin, with propolis showing significant remineralizing and antimicrobial effects.19 Lastly, herb-mediated nanocomposite mouthwashes, such as those using amla seed extract to synthesize graphene oxide–silver nanocomposites, have demonstrated antibacterial and anti-inflammatory effects in the management of plaque-induced gingivitis, offering a novel approach to oral hygiene.20
To address these challenges, nanotechnology-based dental materials have emerged as a promising solution. Incorporating hydroxyapatite (HAp), zirconia (ZrO₂) and silver nanoparticles (AgNPs) into polymer matrices can enhance mechanical strength, impart antibacterial effects and reduce biofilm adhesion. Hydroxyapatite, the primary mineral component of enamel, promotes remineralization and biocompatibility.21, 22 Zirconia improves mechanical durability, while AgNPs provide broad-spectrum antibacterial activity through membrane disruption and reactive oxygen species (ROS) generation.22 This study introduces a novel polystyrene (PS)-based nanocomposite reinforced with HAp, ZrO2 and AgNPs, designed to combine self-cleaning, antibacterial and antiplaque properties. The material has been developed to inhibit bacterial adhesion, disrupt biofilm formation and enhance mechanical performance, offering a multifunctional platform for dental restoration and oral health maintenance.
Material and methods
Chemicals and reagents
All chemicals used in this study were of analytical grade and were employed without further purification, unless otherwise stated. The following materials were employed:
• silver nitrate (AgNO3, ≥99.9%; Sigma-Aldrich, St. Louis, USA);
• trisodium citrate dihydrate (C6H5Na3O7·2H2O, ≥99%; Merck, Darmstadt, Germany);
• zirconium(IV) oxynitrate hydrate (ZrO(NO3)2·xH2O, 99%; Alfa Aesar, Ward Hill, USA);
• citric acid (C6H8O7, ≥99.5%; Sigma-Aldrich);
• calcium nitrate tetrahydrate (Ca(NO3)2·4H2O, ≥99%; Sigma-Aldrich);
• diammonium hydrogen phosphate ((NH4)2HPO4, ≥99%; Merck);
• ammonium hydroxide (NH4OH, 25% w/w in water; Sigma-Aldrich);
• PS pellets (molecular weight ~280,000 g/mol; Sigma-Aldrich);
• toluene (C7H8, ≥99.5%, anhydrous; Sigma-Aldrich);
• Mueller–Hinton agar (MHA; Oxoid, Basingstoke, UK);
• brain heart infusion (BHI) broth (Oxoid);
• crystal violet (C16H18ClN3, ≥90%; Sigma-Aldrich);
• LIVE/DEAD™ BacLight™ Bacterial Viability Kit (Invitrogen, Carlsbad, USA).
Equipment and instrumentation
The following instruments were used for material characterization:
• X-ray diffractometer (D8 Advance; Bruker, Bremen, Germany) – Cu–Kα radiation (λ = 1.5406 Å); 2θ range: 10°–80°; step size: 0.02°; scan speed: 1°/min;
• transmission electron microscope (JEM-2100; JEOL Ltd., Tokyo, Japan) – accelerating voltage: 200 kV; magnification: ×50,000–200,000;
• dynamic light scattering (DLS) and zeta potential analyzer (Zetasizer Nano ZS; Malvern Panalytical, Malvern, UK) – laser wavelength: 633 nm; scattering angle: 173°; temperature: 25°C;
• scanning electron microscope (FEI Quanta 250; FEI Company, Hillsboro, USA) – accelerating voltage: 15 kV; working distance: 10 mm; secondary electron detector;
• atomic force microscope (Dimension Icon; Bruker, Billerica, USA) – tapping mode; scan area: 5 µm × 5 µm; scan rate: 0.5 Hz;
• universal testing machine (Instron 5969; Instron, Norwood, USA) – load cell: 1 kN; crosshead speed: 1 mm/min for flexural tests;
• Vickers hardness tester (MicroMet 5100; Buehler, Lake Bluff, USA) – load: 100 g; dwell time: 15 s;
• contact angle goniometer (ramé-hart 500 Series;
ramé-hart instrument co., Succasunna, USA) – sessile drop method; droplet volume: 5 µL; measurement time: 10 s;
• microplate reader (SpectraMax i3x; Molecular Devices, San Jose, USA) – absorbance measurement at 595 nm;
• confocal laser scanning microscope (LEICA TCS SP8; Leica Microsystems, Wetzlar, Germany) – excitation/emission: 488 nm/500–530 nm for SYTO 9 and 561 nm/575–620 nm for propidium iodide.
Synthesis of nanoparticles
Silver nanoparticles
Silver nanoparticles were synthesized using a chemical reduction method based on the citrate reduction of silver nitrate (AgNO3). Briefly, an aqueous solution of AgNO3 was heated to boiling under vigorous stirring. A trisodium citrate solution was then quickly added to the boiling AgNO3 solution. The color of the solution changed from colorless to pale yellow and then to a characteristic ruby red, indicating the formation of AgNPs. The reaction mixture was maintained at boiling for 30 min until the color stabilized, then allowed to cool to room temperature under continuous stirring. The synthesized AgNPs were characterized using X-ray diffraction (XRD) (D8 Advance), transmission electron microscopy (TEM) (JEM-2100) and DLS (Zetasizer Nano ZS).
Zirconia nanoparticles
Zirconia nanoparticles were synthesized via a sol–gel method using zirconium(IV) oxynitrate hydrate as a precursor and citric acid as a chelating agent. In summary, zirconium oxynitrate was dissolved in deionized water, followed by the addition of citric acid under constant stirring at 60°C to obtain a homogeneous solution. The mixture was then heated to 80°C and stirred until a viscous gel was formed. The gel was dried overnight at 120°C and subsequently calcined at 600°C for 3 h to obtain crystalline ZrO2 nanoparticles. The resulting nanoparticles were analyzed using XRD (D8 Advance) and TEM (JEM-2100).
Hydroxyapatite nanoparticles
Hydroxyapatite nanoparticles were synthesized using a wet chemical precipitation technique. Calcium and phosphate precursors, namely calcium nitrate tetrahydrate and diammonium hydrogen phosphate, were separately dissolved in deionized water. The phosphate solution was then slowly added to the calcium solution under continuous stirring. The pH was maintained at 10–11 by the addition of ammonium hydroxide. The resulting precipitate was aged for 24 h, washed with deionized water and ethanol, and dried at 80°C overnight. The HAp nanoparticles were characterized using XRD (D8 Advance) and TEM (JEM-2100).
Incorporation of nanoparticles into the polystyrene matrix
The incorporation of HAp, ZrO2 and AgNPs into the PS matrix was achieved by solution mixing and a casting process. First, PS pellets were dissolved in toluene (10% w/v) under magnetic stirring at 60°C for 2 h to obtain a homogeneous solution. Separately, the synthesized HAp, ZrO2 and Ag nanoparticles were dispersed in toluene (1 mg/mL) using ultrasonication (40 kHz, 30 min) to ensure uniform dispersion. The nanoparticle suspensions were then added dropwise to the PS solution under constant stirring (500 rpm) at room temperature. The mixture was further stirred for 4 h to ensure thorough mixing and prevent the agglomeration of the nanoparticles. The resulting composite solution was poured into Teflon molds and dried at room temperature in a fume hood for 24 h to allow solvent evaporation. The dried films were subsequently vacuum-dried at 60°C for 12 h to remove residual solvent. The obtained PS–HAp–ZrO2–Ag nanocomposite films were characterized using SEM (FEI Quanta 250) and energy-dispersive X-ray spectroscopy (EDX).
Evaluation of mechanical properties
The mechanical properties of the nanocomposite materials were evaluated in terms of tensile strength, Young’s modulus and elongation at break using a universal testing machine (Instron 5969), in accordance with ASTM D638,23 on dog bone-shaped specimens (gauge length: 25 mm, width: 4 mm, thickness: 2 mm). The hardness of the samples was measured using a Vickers hardness tester (MicroMet 5100), following ISO 6507-1.24 Prior to testing, the samples were polished to a mirror finish, and at least 5 indents were made per sample, with a minimum spacing of 3 diagonal divisions to avoid interaction effects. These tests were conducted to assess the influence of nanoparticle incorporation (HAp, ZrO2, Ag) on the mechanical performance of the PS matrix, which is crucial to assess the suitability and durability of the material in dental applications.
Evaluation of self-cleaning properties
The self-cleaning properties of the nanocomposite surfaces were evaluated by measuring the water contact angle (WCA) using a contact angle goniometer (ramé-hart 500 Series) at room temperature. For each measurement, a 5-µL droplet of deionized water was placed on the surface of the nanocomposite material, and the static contact angle was recorded within 10 s using the sessile drop method. Prior to measurements, the nanocomposite films were cleaned with ethanol and dried under nitrogen flow. At least 5 measurements were taken at different locations on each sample. The average contact angle values were reported together with the standard deviation (SD), which served as a measure of surface wettability and self-cleaning potential.
Evaluation of antibacterial and antiplaque properties
Zone of inhibition assay
Antibacterial activity was primarily tested using the zone of inhibition (ZOI) assay against S. mutans on MHA plates. Sterile paper disks (6-mm diameter, Whatman filter paper) were impregnated with 50 µL of nanocomposite solution (10 mg/mL in tetrahydrofuran (THF)) or small pieces of nanocomposite film (5-mm diameter) for solid samples. The disks were placed on agar plates inoculated with S. mutans at a concentration of 1.5 × 10⁸ CFU/mL (McFarland standard 0.5) and incubated at 37°C for 24 h under aerobic conditions. After incubation, the diameter of the clear zone around the plates (ZOI), which is indicative of antibacterial activity, was measured in millimeters using a digital caliper.
Minimum inhibitory concentration and minimum bactericidal concentration
The minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) were determined using the broth microdilution method in 96-well microplates. Nanocomposite extracts were prepared by immersing nanocomposite films (10 mg/mL) in sterile BHI broth for 24 h at 37°C with shaking. These extracts, along with serial twofold dilutions of AgNP solutions (ranging from 0.78 ppm to 100 ppm), were serially diluted in BHI broth in the microplates. A bacterial suspension of S. mutans adjusted to 5 × 105 CFU/mL was added to each well (100 µL bacterial suspension + 100 µL diluted extract/AgNPs). The plates were incubated at 37°C for 24 h. The MBC was determined visually as the lowest concentration of nanocomposite extract or AgNPs that inhibited visible bacterial growth, as indicated by the absence of turbidity in the wells.
Colony-forming unit assay
Quantitative antibacterial efficacy of the nanocomposite was evaluated using a colony-forming unit (CFU) assay. The nanocomposite films (1 cm × 1 cm) were sterilized under ultraviolet (UV) light for 30 min before testing. The films were then immersed in 1 mL of S. mutans bacterial suspension at a concentration of 1 × 106 CFU/mL in BHI broth for different time intervals (1, 3, 6, and 24 h) at 37°C. At each time point, 100 µL of the bacterial suspension was serially diluted in sterile phosphate-buffered saline (PBS), and 100 µL of each dilution was plated in triplicate on BHI agar plates. After incubation at 37°C for 24 h, colonies were counted manually, and the percentage of bacterial reduction compared to the control group (bacteria incubated without nanocomposite films) was calculated.
Antiplaque biofilm assay
The formation of S. mutans biofilms on nanocomposite films was evaluated using the crystal violet assay. Sterile nanocomposite films (1 cm × 1 cm) were placed in 24-well plates and inoculated with 1 mL of S. mutans suspension (1 × 106 CFU/mL) in BHI broth supplemented with 1% sucrose (Sigma-Aldrich) to promote biofilm formation for 24, 48 and 72 h at 37°C under static conditions. After incubation, the planktonic bacteria were carefully aspirated, and the biofilms were gently washed twice with 1 mL of PBS (pH 7.4). The biofilms were then fixed with 500 µL of absolute methanol per well for 20 min. After fixation, methanol was removed and the biofilms were stained with 500 µL of crystal violet solution (0.1% w/v in water) per well for 30 min. The excess stain was removed by washing the wells 3 times with 1 mL of PBS. The dye bound to the biofilm was then dissolved by adding 500 µL of 33% acetic acid per well and incubating at room temperature for 30 min. Finally, 200 µL of the dissolved dye solution from each well was transferred to a 96-well microplate, and the absorbance was measured at 595 nm using a microplate reader (SpectraMax i3x). Biofilm biomass was quantified based on the absorbance values, with higher absorbance indicating greater biofilm formation.
Confocal laser scanning microscopy
A confocal laser scanning microscope (LEICA TCS SP8) was used for detailed visualization of the biofilms. The S. mutans biofilms were grown on nanocomposite films for 48 h, as described above. After incubation, the biofilms were carefully washed with PBS and stained using the LIVE/DEAD™ BacLight™ Bacterial Viability Kit according to the manufacturer’s protocol. SYTO 9 (green fluorescence) stains viable cells, whereas propidium iodide (red fluorescence) labels dead bacteria. The imaging of the biofilms was conducted using confocal laser scanning microscopy (CLSM) with excitation/emission wavelengths of 488 nm/500–530 nm for SYTO 9 and 561 nm/575–620 nm for propidium iodide. Z-stack images were taken at 1-µm intervals. The thickness and structure of the biofilm, including the distribution of live and dead bacteria, were analyzed using the ImageJ software (https://imagej.net/ij/download.html).
Statistical analysis
The quantitative data was expressed as mean ±SD from at least 3 independent experiments. Statistical analysis was performed using one-way analysis of variance (ANOVA) for comparisons involving more than 2 groups, or Student’s t-test for comparisons between 2 groups. When ANOVA indicated significant differences, Tukey’s honestly significant difference (HSD) post hoc test was applied to perform pairwise comparisons and determine which groups were significantly different from each other. The analyses were conducted using the GraphPad Prism v. 9.0 software (GraphPad Software, San Diego, USA). A p-value <0.05 was considered statistically significant.
Results
Synthesis of silver nanoparticles
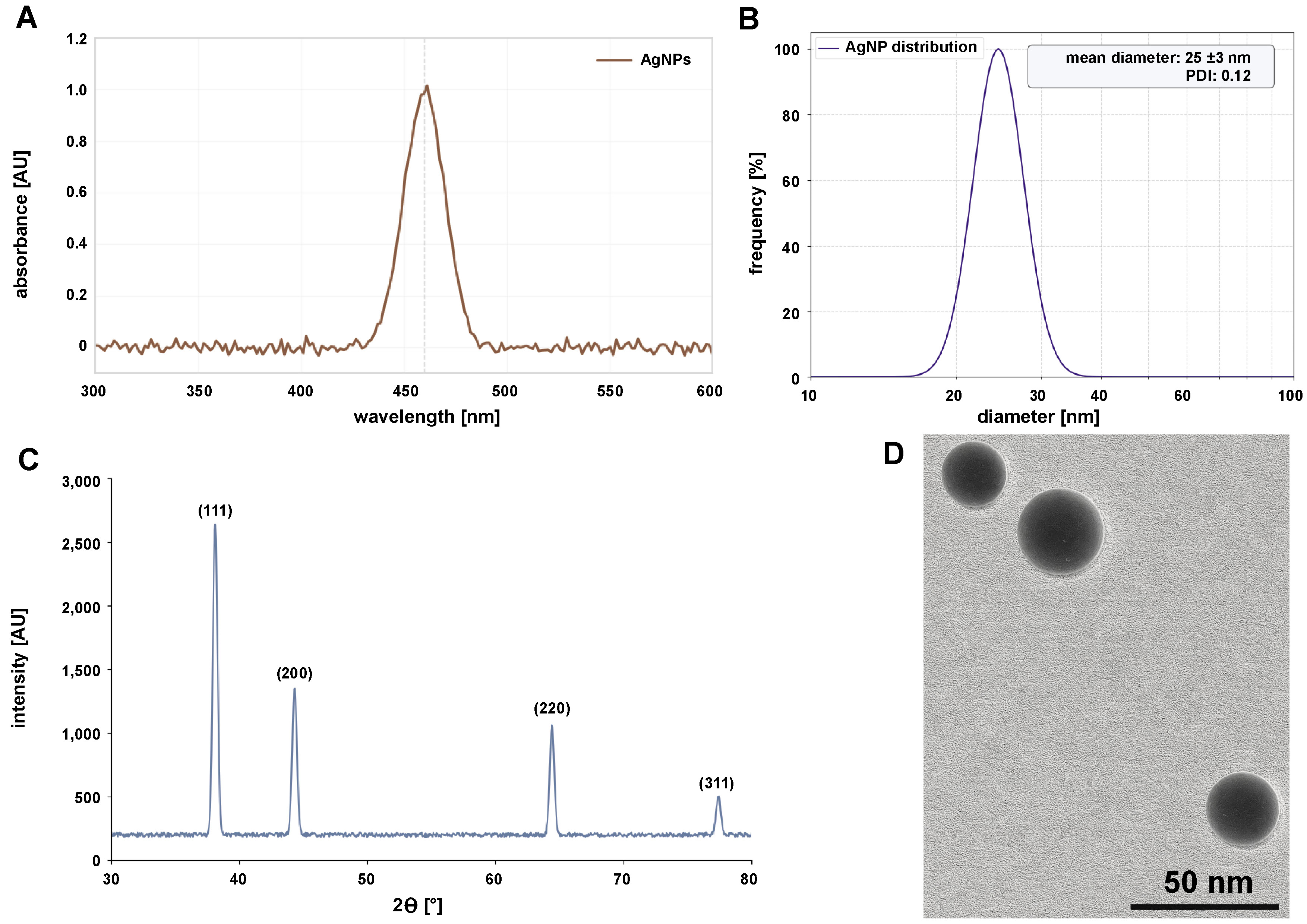
Silver nanoparticles were successfully synthesized via a chemical reduction method using trisodium citrate as a reducing and stabilizing agent. Within 5 min of citrate addition, the initially colorless AgNO3 solution turned pale yellow, indicating the nucleation of AgNPs. After 15 min of boiling, the solution exhibited a characteristic ruby-red color, confirming the formation of spherical AgNPs due to surface plasmon resonance (SPR). The deep red color remained stable after 30 min of boiling, indicating complete reduction and stabilization of the nanoparticles. The synthesized AgNPs showed a strong SPR peak at 460 nm, characteristic of spherical AgNPs with a diameter of 20–30 nm (Figure 2A). The narrow peak width indicated a monodisperse size distribution. The hydrodynamic diameter of the AgNPs was 25 ±3 nm with a polydispersity index (PDI) of 0.12, confirming uniformity (Figure 2B). The zeta potential was −30 ±2 mV, indicating high colloidal stability due to electrostatic repulsion imparted by the citrate cap. The nanoparticles were well dispersed, with minimal aggregation observed. The XRD analysis exhibited 4 distinct peaks at 2θ values of 38.1°, 44.3°, 64.4°, and 77.4°, corresponding to the (111), (200), (220), and (311) crystallographic planes of face-centered cubic (FCC) silver (JCPDS No. 04-0783) (Figure 2C). The crystallite size, calculated using the Scherrer equation, was 18 nm, which is consistent with TEM observations. Transmission electron micrographs confirmed the formation of spherical AgNPs with an average diameter of 22 ±4 nm (Figure 2D).
Synthesis of zirconia nanoparticles
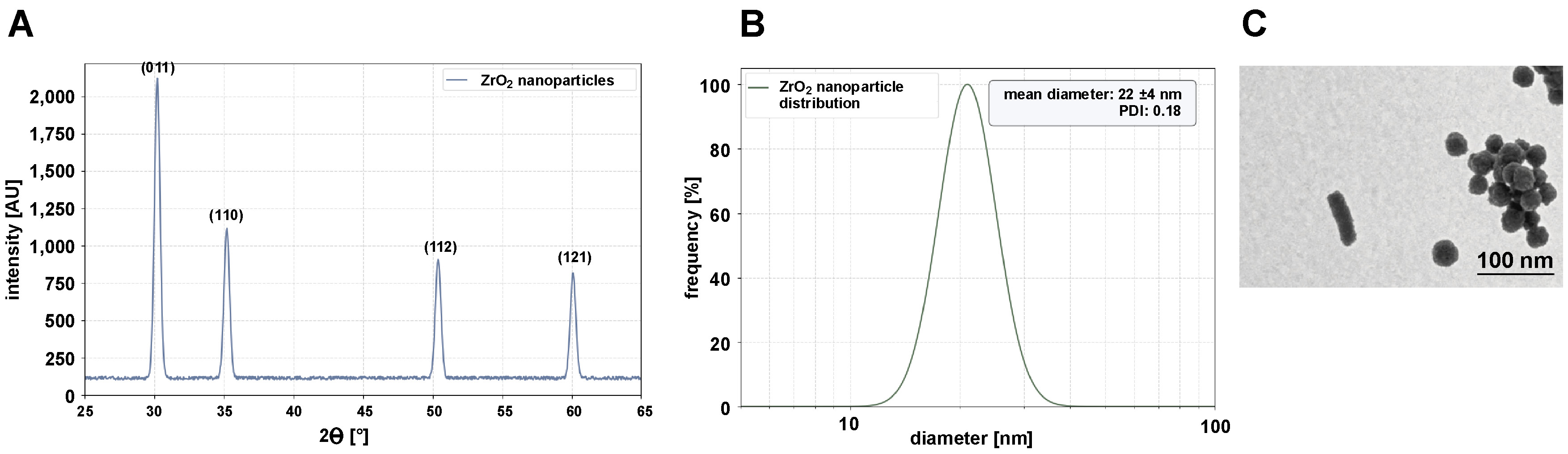
Zirconia nanoparticles were successfully synthesized via a sol–gel method using zirconium(IV) oxynitrate hydrate as a precursor and citric acid as a chelating agent. The process involved controlled gelation, drying and calcination to produce crystalline ZrO2 nanoparticles. The addition of citric acid to the zirconium oxynitrate solution resulted in a clear, homogeneous mixture after 30 min of stirring at 60°C. When heated to 80°C, the solution gradually transformed into a viscous, translucent gel within 4 h, indicating the formation of a zirconium citrate complex. The gel was dried overnight at 120°C, resulting in a brittle, yellowish xerogel. Calcination at 600°C for 3 h yielded a white crystalline powder, confirming the removal of organic residues and the formation of ZrO2. The XRD analysis revealed sharp diffraction peaks at 2θ values of 30.2°, 35.2°, 50.4°, and 60.1°, corresponding to the (011), (110), (112), and (121) planes of tetragonal zirconia (JCPDS No. 17-0923) (Figure 3A). The absence of impurity peaks confirmed the high purity of the synthesized ZrO2. The crystallite size, calculated according to the Scherrer equation, was 15 nm. The nanoparticles exhibited low agglomeration, with clear lattice fringes (d-spacing = 0.29 nm) corresponding to the (011) plane of tetragonal ZrO2. The hydrodynamic diameter of ZrO2 nanoparticles in aqueous suspension was 22 ±4 nm with a PDI of 0.18, indicating a narrow size distribution. The zeta potential was −25 ±1 mV, denoting moderate colloidal stability due to residual citrate ions on the surface (Figure 3B). Transmission electron micrographs showed spherical to quasi-spherical morphology with an average particle size of 18 ±3 nm (Figure 3C).
Synthesis of hydroxyapatite nanoparticles
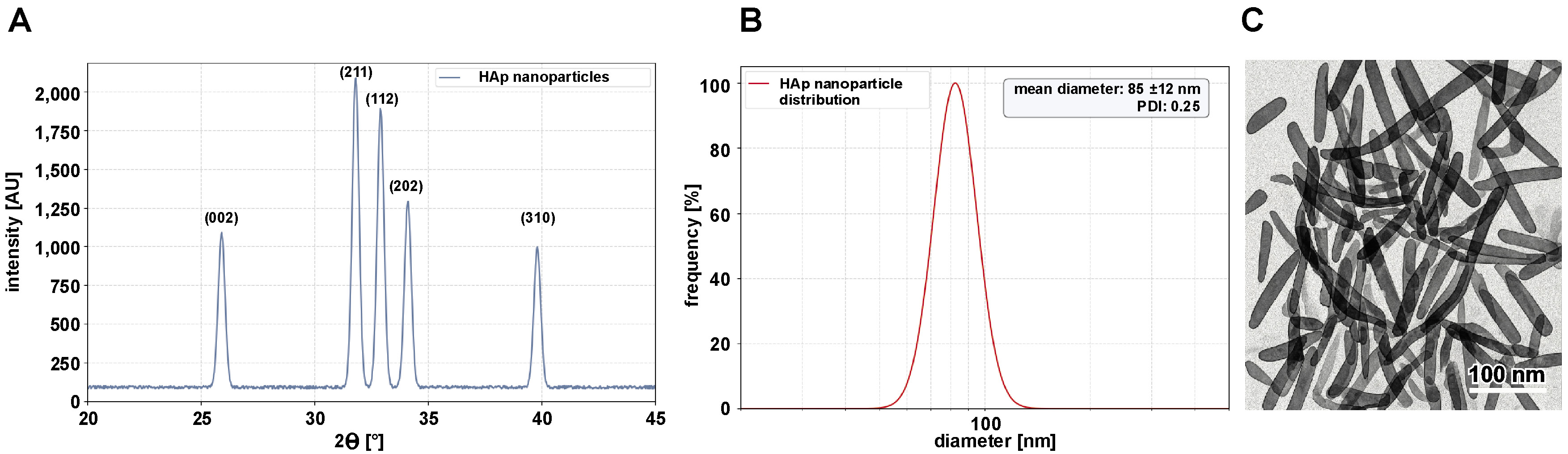
Hydroxyapatite nanoparticles were successfully synthesized using a wet chemical precipitation method with calcium nitrate tetrahydrate and diammonium hydrogen phosphate as calcium and phosphate precursors, respectively. The reaction was carried out at a controlled pH of 10–11 to ensure the formation of stoichiometric HAp. After adding the phosphate solution dropwise to the calcium solution, a milky white precipitate formed immediately, indicating nucleation. The reaction mixture was stored at room temperature for 24 h to promote crystallinity and particle growth. The precipitate was washed 3 times with deionized water and ethanol via centrifugation (5,000 rpm, 10 min) to remove residual ammonium and nitrate ions. After drying overnight at 80°C, a fine white powder was obtained. The XRD analysis confirmed the formation of phase-pure HAp, with characteristic peaks at 2θ values of 25.9°, 31.8°, 32.9°, 34.1°, and 39.8°, corresponding to the (002), (211), (112), (202), and (310) planes of hexagonal HAp, respectively (JCPDS No. 09-432) (Figure 4A). The sharp, well-defined peaks indicate high crystallinity, with a crystallite size of 30 nm, as calculated using the Scherrer equation. The hydrodynamic diameter of the HAp nanoparticles in aqueous suspension was 85 ±12 nm with a PDI of 0.25, indicating slight agglomeration in the solution (Figure 4B). The zeta potential was −20 ±2 mV, suggesting moderate colloidal stability due to the presence of hydroxyl groups on the surface. Transmission electron micrographs revealed rod-shaped HAp nanoparticles with an average length of 60 ±10 nm and a diameter of 20 ±5 nm. The nanoparticles exhibited uniform morphology with minimal aggregation, consistent with the controlled pH and aging process (Figure 4C).
Characterization of the PS–HAp–ZrO2–Ag nanocomposite
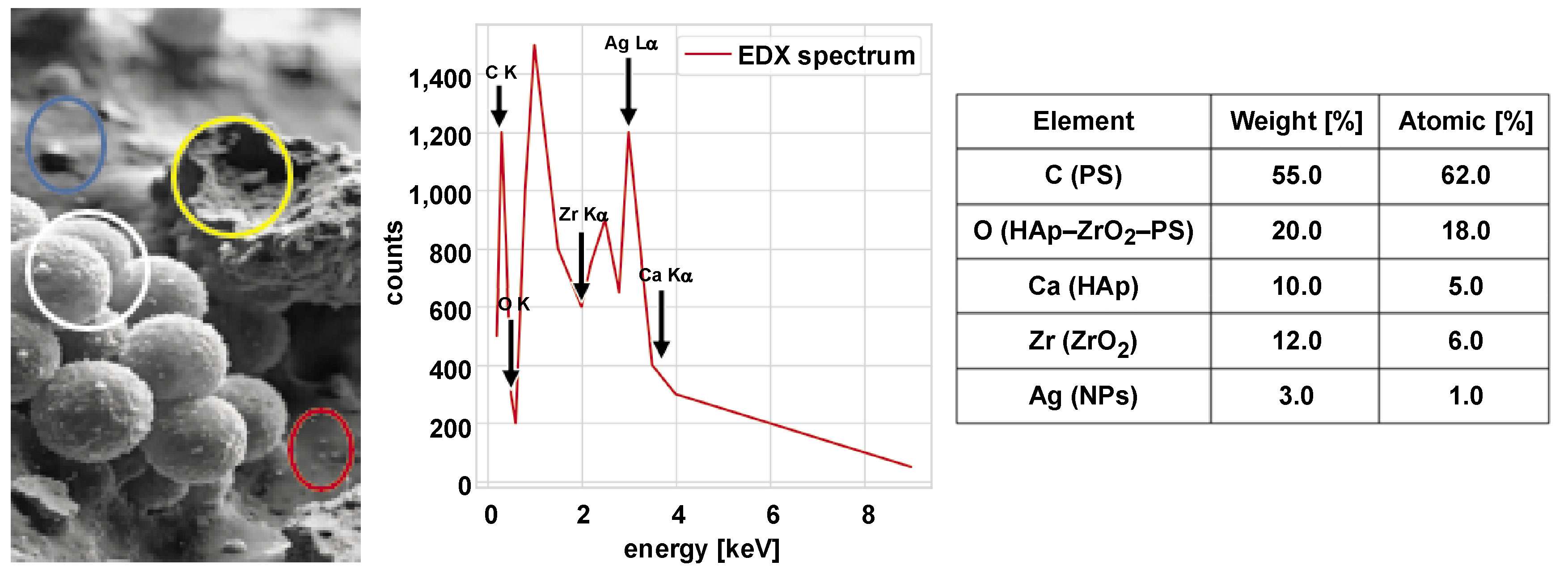
The PS–HAp–ZrO2–Ag nanocomposite was successfully prepared by mixing and pouring a solution, which ensured uniform dispersion of HAp, ZrO2 and AgNPs within the PS matrix. The PS solution in toluene (10% w/v) remained clear and homogeneous after 2 h of stirring at 60°C. The suspensions of nanoparticles (HAp, ZrO2, Ag) in toluene showed no visible sedimentation after ultrasonication, indicating effective dispersion. After dropwise addition of the nanoparticle suspensions to the PS solution, the mixture retained a uniform, slightly opaque appearance, suggesting good compatibility between the nanoparticles and the polymer matrix. The composite solution was cast into Teflon molds, yielding flexible, free-standing films after 24 h of solvent evaporation. Subsequent vacuum drying at 60°C for 12 h resulted in smooth, crack-free films with a thickness of 200 ±10 µm. Scanning electron microscopy images revealed a homogeneous surface morphology without visible nanoparticle agglomeration. At higher magnification, well-dispersed nanoparticles embedded in the PS matrix were observed, with HAp rods and spherical ZrO2 and AgNPs. The EDX mapping confirmed the uniform distribution of Ca (HAp), Zr (ZrO2) and Ag (AgNPs) across the composite surface (Figure 5).
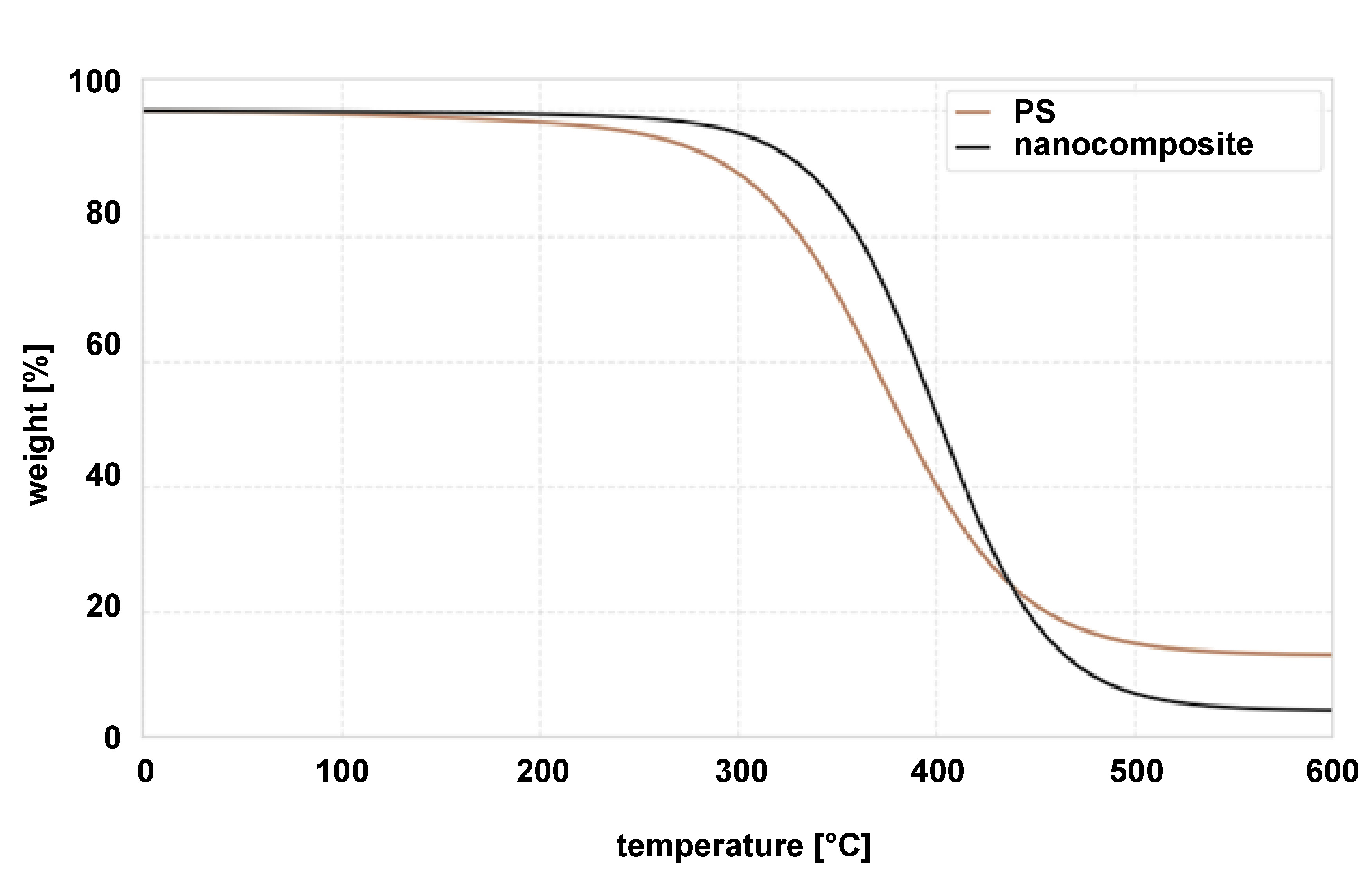
Thermogravimetric analysis (TGA) of pure PS and the PS–HAp–ZrO₂–Ag nanocomposite is presented in Figure 6. The TGA curve for pure PS shows a single degradation step, with onset at around 350°C and significant weight loss between 400°C and 450°C, which corresponds to the decomposition temperature of PS. The nanocomposite displayed a similar decomposition step within the same temperature range (400–450°C), indicating that the incorporation of nanoparticles did not significantly change the thermal decomposition characteristics of the PS matrix. However, the residual mass of the nanocomposite at 600°C is 5.2%, closely matching the theoretical nanoparticle loading (5 wt%). This result confirms the successful incorporation of HAp, ZrO2 and Ag nanoparticles into the PS matrix. A slight variation in the onset of degradation between the pure PS and the nanocomposite could be attributed to the presence of the nanoparticles, which may slightly affect the thermal stability of the polymer matrix. Nevertheless, the overall thermal decomposition profile remained largely unchanged, indicating that the nanoparticles were well dispersed and did not significantly interfere with the thermal degradation process of PS. The consistency between the experimental residual mass and the theoretical loading confirms the synthesis and composition of the nanocomposite.
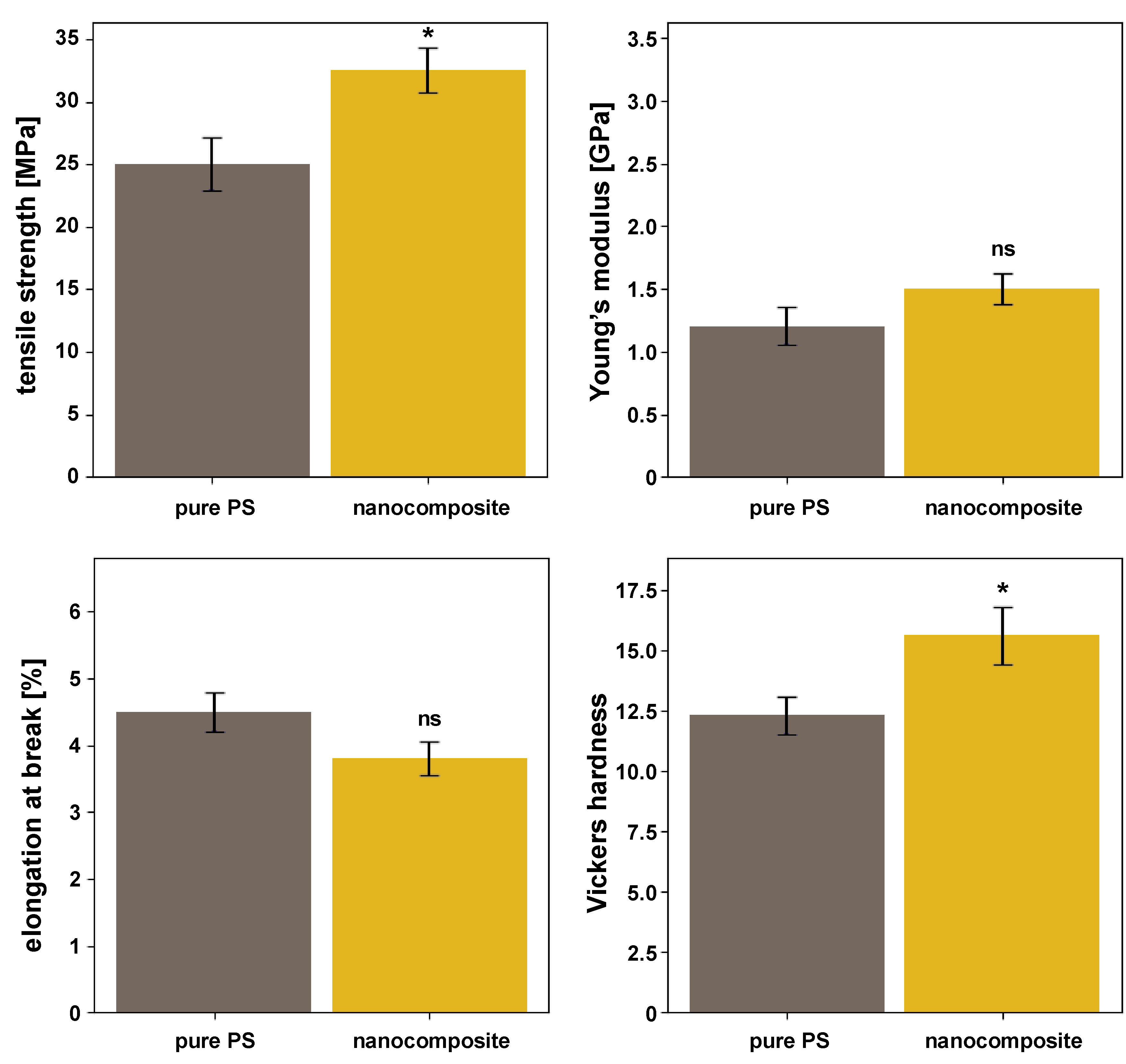
The mechanical properties of pure PS and the PS–HAp–ZrO2–Ag nanocomposite were evaluated using tensile testing and Vickers hardness measurements. Tensile testing revealed a significant improvement in the mechanical strength of the nanocomposite. The tensile strength increased by 30% (from 25.0 ±2.1 MPa for pure PS to 32.5 ±1.8 MPa for the nanocomposite; p < 0.01), indicating a reinforcing effect due to the incorporation of nanoparticles. Similarly, the Young’s modulus increased by 25% (from 1.2 ±0.15 GPa to 1.5 ±0.12 GPa), although the difference was not statistically significant. However, the elongation at break decreased slightly by 15.5% (from 4.5 ±0.3% to 3.8 ±0.25%; p = 0.07), consistent with the expected trade-off between stiffness and ductility due to the addition of rigid nanoparticles. Vickers hardness measurements further confirmed mechanical reinforcement. The nanocomposite exhibited a significant increase in hardness compared to pure PS. The Vickers hardness number (HV) for the nanocomposite was 15.6 ±1.2, whereas pure PS had a hardness of 12.3 ±0.8 (p < 0.05), corresponding to an approximate 27% increase. This improvement is attributed to the high hardness of the ceramic nanoparticles (HAp and ZrO2), which enhanced the overall resistance of the nanocomposite to indentation (Figure 7).
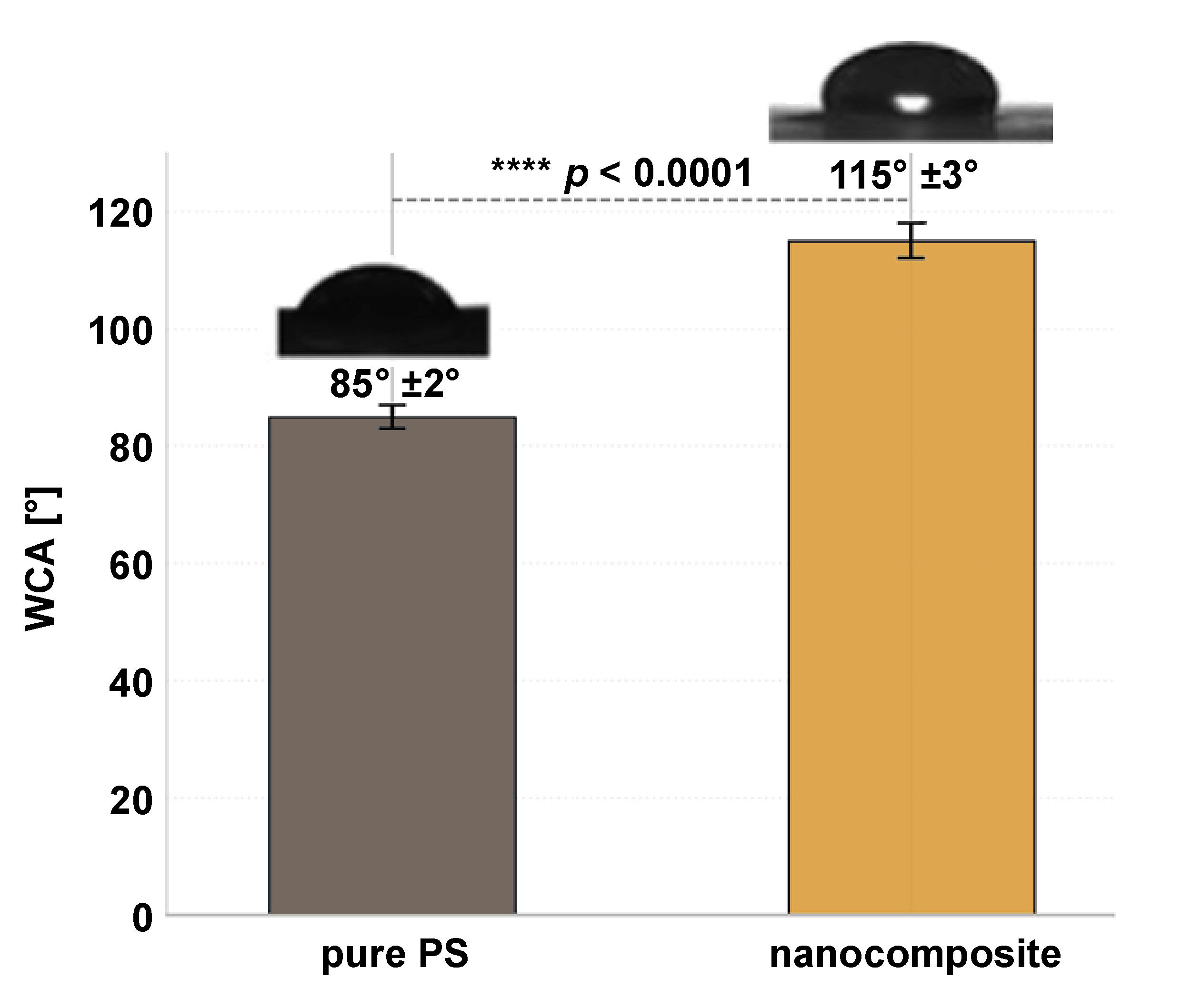
The self-cleaning properties of the PS–HAp–ZrO2–Ag nanocomposite were evaluated by measuring the WCA using the sessile drop method. Pure PS exhibited a WCA of 85° ±2°, indicating moderate hydrophobicity due to its non-polar nature. In contrast, the nanocomposite showed a significantly higher WCA of 115° ±3°, reflecting a marked improvement in hydrophobicity compared to pure PS (p < 0.001) (Figure 8). This increase in water repellency can be attributed to the combined effects of low surface energy (from ZrO2 and AgNPs) and surface micro/nanoroughness, which may promote air trapping and reduce the contact area between water droplets and the solid surface. While the surface exhibited low water adhesion and a beading effect, it did not meet the criterion for superhydrophobicity (WCA > 150°). Nevertheless, the increased hydrophobicity contributes to reduced bacterial adhesion and improved self-cleaning potential, as water droplets roll off more easily, carrying away contaminants such as bacteria and organic debris.
Results of antibacterial activity assessment
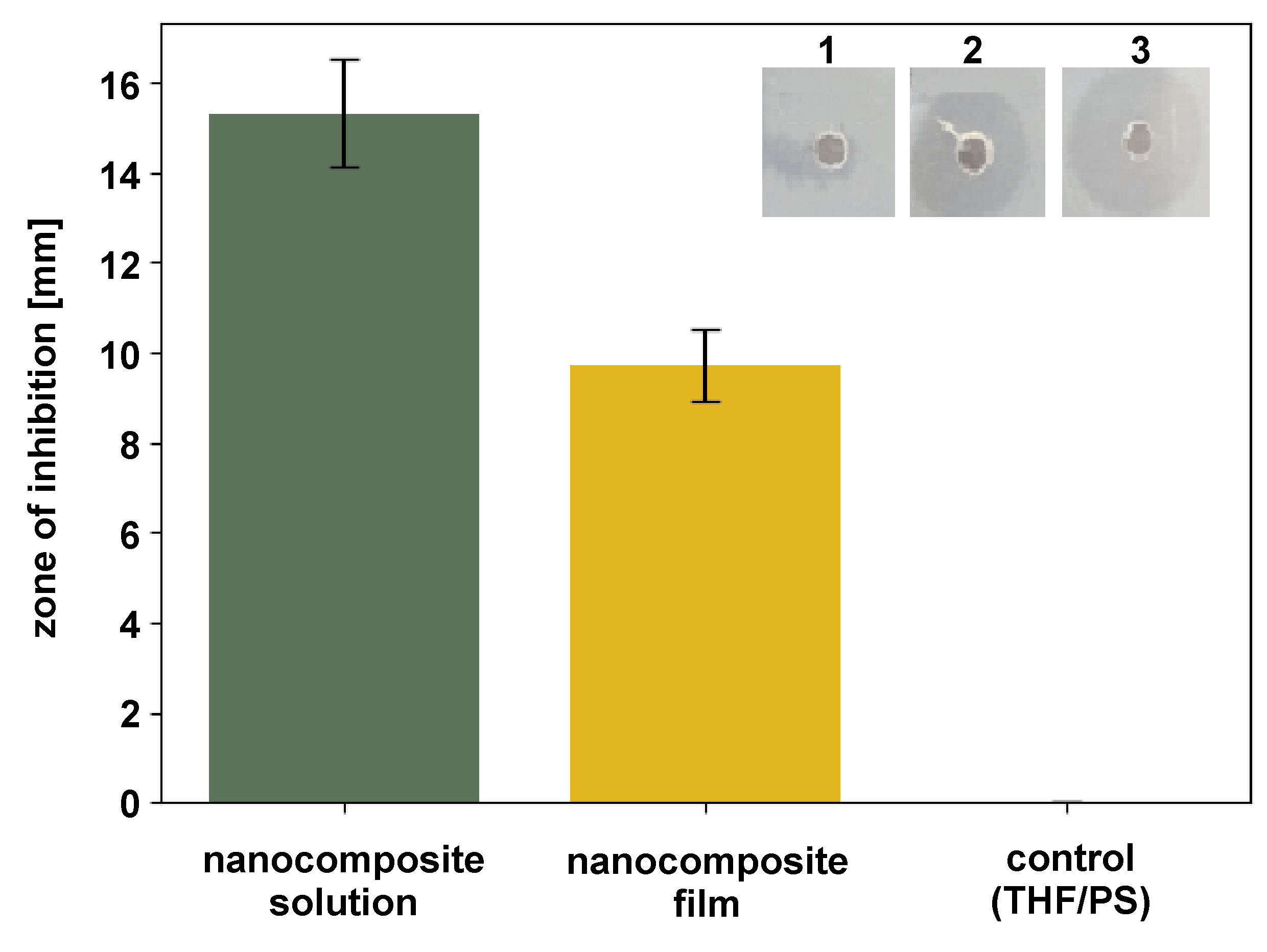
The ZOI assay demonstrated significant antibacterial activity of the PS–Hap–ZrO2–Ag nanocomposite against S. mutans (Figure 9). For the nanocomposite solution (10 mg/mL in THF), the impregnated disks produced a mean inhibition zone of 15.3 ±1.2 mm (n = 3), whereas solid nanocomposite films (5-mm diameter) produced a smaller zone of 9.7 ±0.8 mm (n = 3). These results confirm the release of bioactive agents likely due to the presence of AgNPs, which effectively inhibited bacterial growth around the test samples.
Both ZOI values exceeded the threshold for clinically relevant antibacterial activity (>6 mm), highlighting the potential of the nanocomposites to interfere with S. mutans proliferation. The larger inhibition zones observed for solution-based samples compared to solid films suggest improved diffusion of antibacterial components in the liquid phase. Control samples (THF-impregnated disks or pristine PS films) showed no inhibition (0 mm), confirming that the antibacterial effect is intrinsic to the nanocomposite formulation.
These results confirm the successful incorporation of AgNPs into the HAp–ZrO2–PS matrix and emphasize their crucial role in conferring antibacterial properties relevant for dental applications.
Minimum inhibitory concentration and minimum bactericidal concentration
Both the nanocomposite extract and AgNPs exhibited strong antibacterial activity against S. mutans (Table 1). The MIC was 6.25 mg/mL for the nanocomposite extract and 3.12 ppm for the AgNPs, indicating a strong inhibitory effect. The MBC values were twice as high as the MIC values (12.5 mg/mL for the nanocomposite and 6.25 ppm for the AgNPs), confirming the bactericidal effect. No visible bacterial growth was observed at these concentrations, and bacterial viability was reduced by ≥99.9%.
Colony-forming unit assay
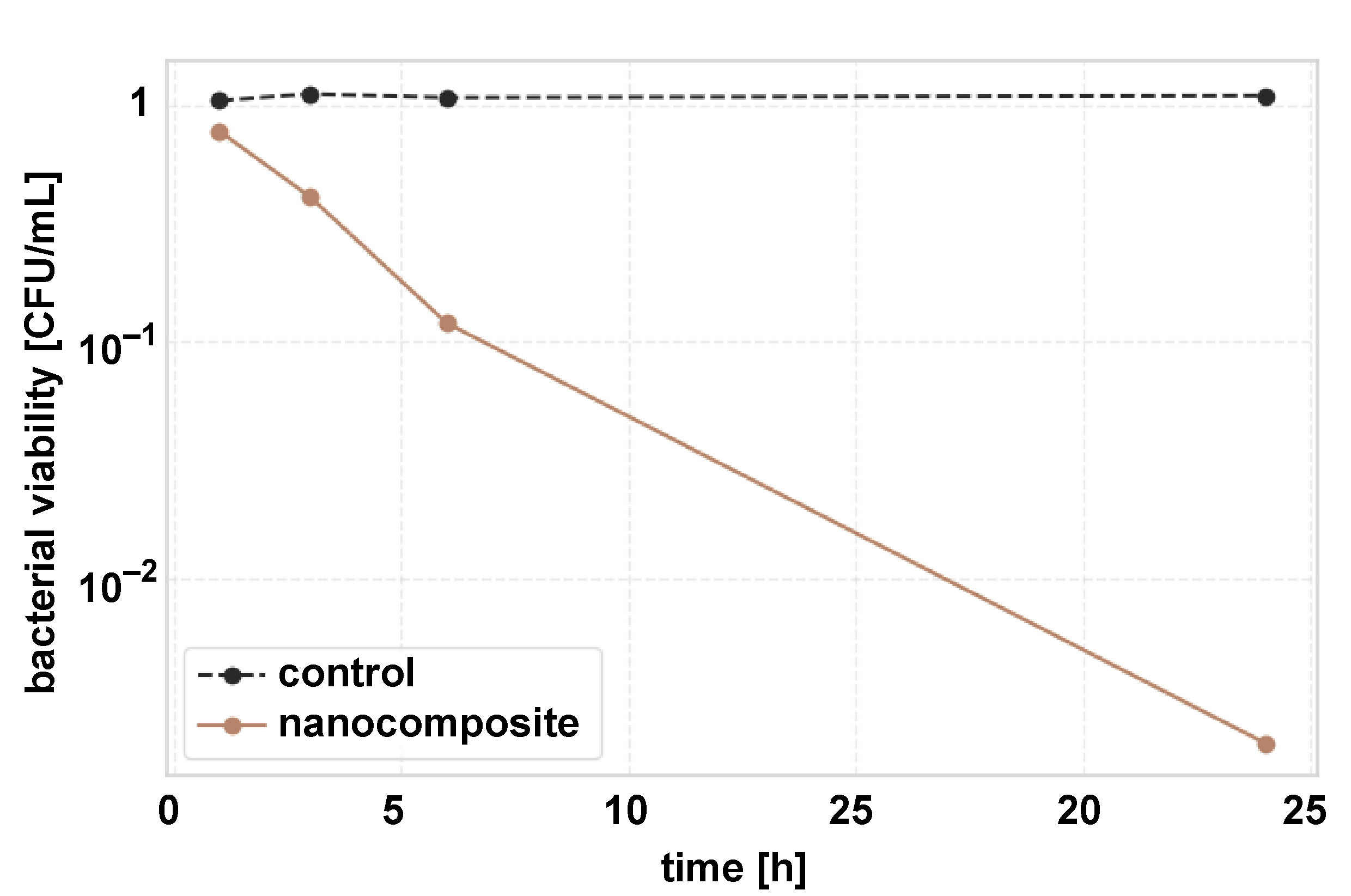
The nanocomposite films exhibited time-dependent antibacterial activity (Table 2). A reduction in bacterial count was observed after 1 h (25.4%) and increased progressively. After 24 h, a 99.8% reduction in CFU/mL was observed compared to the control. Complete inhibition (>99.9%) was achieved at AgNP concentrations ≥6.25 ppm.
Time–kill kinetics of the nanocomposite films against S. mutans was also investigated using the CFU assay (Figure 10). A rapid and exponential reduction in bacterial viability was observed over 24 h. While the control samples maintained a stable bacterial count (1.05 ±0.12 × 106 CFU/mL to 1.10 ±0.13 × 106 CFU/mL), the nanocomposite-treated samples showed a time-dependent bactericidal effect. The semi-logarithmic plot illustrates the ability of the nanocomposite to achieve >3-log reduction (99.9% elimination) within 24 h (p < 0.001, one-way ANOVA) (Figure 10).
Antibiofilm assay
Biofilm formation
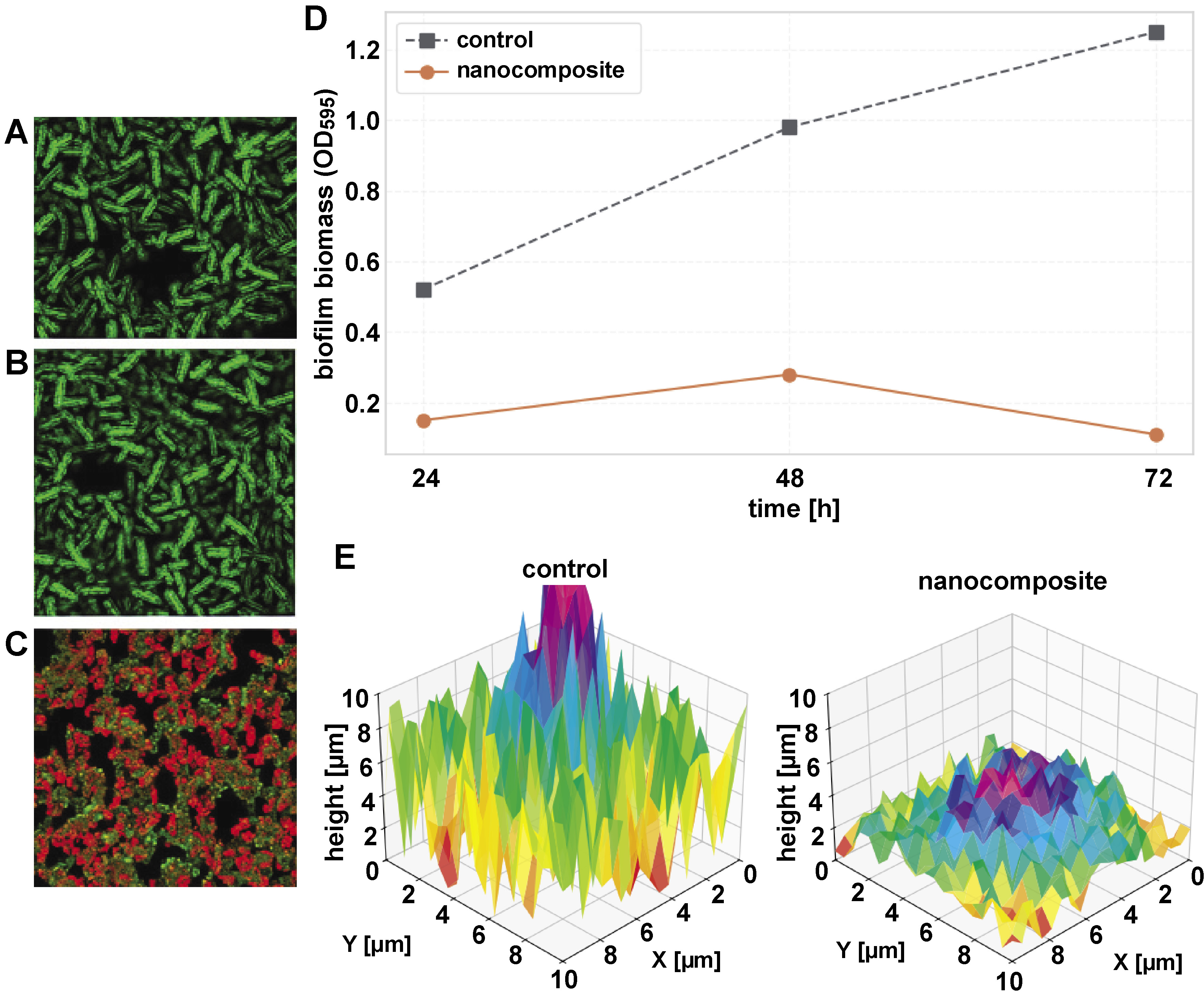
The nanocomposite films significantly inhibited the biofilm formation of S. mutans in a time-dependent manner (Table 3). Crystal violet staining showed a 75.6% reduction in biofilm biomass after 24 h compared to the PS control (p < 0.001). After 72 h, biofilm inhibition increased to 91.2% (p < 0.001), with absorbance values decreasing from 1.25 ±0.15 (control) to 0.11 ±0.02 (nanocomposite).
Confocal laser scanning microscopy
Confocal laser scanning microscopy showed significant disruption of S. mutans biofilm architecture on nanocomposite surfaces. Quantitative analysis showed a 74.8% reduction in biofilm thickness, decreasing from 32.5 ±3.1 µm (control) to 8.2 ±1.5 µm after 48 h of incubation. The live/dead cell ratio shifted markedly from 4.7:1 (predominantly viable cells in control biofilms) to 0.6:1 (predominantly dead cells on nanocomposite surfaces), confirming strong bactericidal activity rather than mere growth inhibition. Structurally, control samples exhibited dense, multilayered biofilms with nearly complete surface coverage, whereas nanocomposite-treated surfaces showed fragmented microcolonies and sparse colonization covering less than 20% of the surface. The control biofilms demonstrated high green fluorescence due to the dominance of live S. mutans cells, with minimal red fluorescence from dead cells. This reflects the natural progression of biofilm maturation, where viable cells dominate the structure (Figure 11A–C). These results emphasize the dual antiplaque mechanism of the nanocomposite: direct killing of bacteria and physical disruption of biofilm maturation. The time-dependent antibiofilm efficacy of the HAp–ZrO2–PS nanocomposite was quantitatively evaluated using crystal violet staining (Figure 11D). Across all evaluated time points, the biofilm biomass values of the nanocomposite-treated group remained significantly lower than those of the PS control group, with absorbance values decreasing by 75.6% (p < 0.001) at 24 h and by 91.2% (p < 0.001) at 72 h compared to the PS control. The nanocomposite group showed minimal biofilm formation (optical density (OD)595 = 0.11 ±0.02), while the control samples showed progressive biofilm accumulation after 72 h (OD595 = 1.25 ±0.15). To better visualize the biofilm architecture, three-dimensional (3D) reconstructions were generated from CLSM Z-stack images. Figure 11E shows the 3D representation of S. mutans biofilms on control and PS–HAp–ZrO2–Ag nanocomposite surfaces. The control samples exhibited a dense, multilayered biofilm structure with high vertical variation, indicating thick and mature biofilm formation. In contrast, the nanocomposite surfaces showed significantly thinner, discontinuous structures with reduced vertical height and sparse bacterial clusters, reflecting the material’s ability to inhibit biofilm maturation and promote bacterial cell death. These results demonstrate the ability of the nanocomposite to suppress both the initial bacterial adhesion and the long-term maturation of the biofilm.
Discussion
This study demonstrates a novel approach to the development of multifunctional dental nanocomposites based on a PS matrix enriched with HAp, ZrO2 and AgNPs. The main objective was to design a material exhibiting self-cleaning, antibacterial and antiplaque properties that utilizes the synergistic effects of these nanoscale components. The results confirm the successful synthesis of individual nanoparticles, their effective incorporation into the PS matrix and the achievement of the desired functionalities.
The successful synthesis of well-defined nanoparticles is of fundamental importance for the performance of the final nanocomposite. The chemical reduction method yielded spherical AgNPs with an average size of approx. 22 nm (TEM) and a hydrodynamic diameter of 25 nm (DLS), exhibiting a characteristic SPR peak at 460 nm and high colloidal stability (−30 mV zeta potential).25, 26, 27 These results are consistent with previous reports on citrate-stablized AgNPs, which typically produce spherical nanoparticles in the 10–50 nm range with strong SPR absorption (420–460 nm) and good stability due to citrate capping.28, 29, 30, 31 The narrow size distribution (PDI = 0.12) and confirmed FCC structure (XRD) further validate the synthesis protocol. The small particle size and high surface area are crucial for maximizing the antibacterial efficacy of AgNPs.32
Similarly, the sol–gel synthesis produced relatively monodisperse tetragonal ZrO2 nanoparticles (TEM: ~18 nm; DLS: 22 nm; PDI = 0.18). The tetragonal phase, confirmed by XRD, is known for its superior mechanical properties compared to the monoclinic phase, making it desirable for reinforcement applications.33, 34 The particle size is consistent with sol–gel methods where calcination occurs at moderate temperatures (e.g., 600°C).35 The moderate zeta potential (−25 mV) indicates adequate dispersion stability, which is supported by residual citrate from the synthesis process.
The wet precipitation method yielded phase-pure, crystalline, rod-shaped HAp nanoparticles (approx. 60 nm × 20 nm), corresponding to the targeted hexagonal structure (JCPDS No. 09-432). This morphology is commonly observed in HAp synthesized under controlled pH and aging conditions.36 While well-defined rods were noted in TEM, the larger hydrodynamic diameter (85 nm) and higher PDI (0.25) in DLS indicate some degree of agglomeration in aqueous suspension, a known challenge for HAp nanoparticles.37 Nevertheless, the successful synthesis of nanoscale HAp, a biocompatible and osteoconductive material structurally similar to dental enamel, is essential for potential dental applications. Moreover, it contributes to the biointegration and mechanical strength of the composite.38
Solution mixing followed by solvent casting proved effective for the fabrication of homogeneous PS–HAp–ZrO2–Ag nanocomposites. Scanning electron microscopy imaging in conjunction with EDX mapping confirmed uniform distribution of all nanoparticle components within the PS matrix, which is crucial for achieving uniform properties of the material.39 The absence of large agglomerates minimizes stress concentration and ensures optimal load transfer and surface properties.40
The TGA results showed that the incorporation of 5 wt% nanoparticles did not significantly alter the onset of thermal degradation of the PS matrix, indicating good interfacial compatibility and no catalytic degradation by the nanoparticles at these temperatures. The measured residual mass corresponded to the theoretical loading, confirming the successful incorporation of the inorganic components.41 The preserved thermal stability is sufficient for many dental processing techniques and intraoral conditions.
Mechanical testing demonstrated a significant improvement in material performance. The tensile strength increased by 30%, while the modulus of elasticity showed a 25% increase compared to pure PS. This enhancement is attributed to the high stiffness of the ceramic HAp and ZrO2 nanoparticles and effective stress transfer from the polymer matrix to the well-dispersed fillers.42, 43 The rod-like morphology of HAp may further contribute to reinforcement.44 The slight decrease in elongation at break reflects a typical trade-off, where rigid fillers are incorporated into a polymer matrix, indicating increased stiffness and reduced extensibility. These improved mechanical properties are advantageous for dental materials that require durability and wear resistance.45
An important innovation of this study is the improvement in self-cleaning properties. The surface of the nanocomposite showed a significantly increased hydrophobicity (WCA = 115°) compared to moderately hydrophobic pure PS (WCA = 85°). Although this value does not meet the criterion for superhydrophobicity (>150°), it indicates a clear shift toward a low-energy, water-repellent surface. This behavior is likely due to the combined influence of the low surface energy ZrO2 nanoparticles and the AgNPs forming a hierarchical micro/nanostructure on the surface.46, 47 The induced roughness can trap air pockets (Cassie–Baxter state) and minimize the contact area between water droplets and the solid surface.48 Although HAp is generally hydrophilic, its nanoscale dispersion and potential partial coverage by the hydrophobic matrix or other nanoparticles seem to prevent it from dominating the surface wettability. Increased hydrophobicity is closely related to reduced bacterial adhesion and easier removal of impurities, which is beneficial for limiting plaque accumulation.49, 50 Similar increases in hydrophobicity have been reported for polymer composites containing ZrO2 or other nanoparticles designed for reduced fouling.51, 52
The incorporation of AgNPs conferred significant antibacterial activity to the nanocomposite, as shown in several tests with S. mutans, a major causative agent of dental caries.53 The ZOI assay confirmed the release of active compounds from both the dissolved and solid forms of the nanocomposite, with inhibition zones exceeding clinically relevant thresholds.54 The larger zones observed for solution-based samples indicate easier diffusion of Ag+ ions or nanoparticles.55
The MIC and MBC results quantified this potency and showed strong inhibitory (MIC = 6.25 mg/mL for extract, MIC = 3.12 ppm for AgNPs) and bactericidal (MBC = 12.5 mg/mL for extract, MBC = 6.25 ppm for AgNPs) effects. These values are comparable to or better than those reported for other AgNP-based dental materials.56, 57 The twofold difference between MIC and MBC values supports a bactericidal mechanism rather than just bacteriostatic activity. The CFU assay further corroborated these results, showing a time-dependent reduction in bacterial viability, achieving >3-log reduction (>99.9%) within 24 h. This rapid bactericidal effect is attributed to the release of Ag+ ions, which destroy bacterial cell membranes, impair metabolic enzymes and trigger oxidative stress via ROS formation.22, 58, 59
In addition to killing planktonic bacteria, the nanocomposite exhibited significant efficacy in preventing and disrupting S. mutans biofilms, which are the predominant form of bacterial growth in the oral cavity and are notoriously difficult to eradicate. Crystal violet assays showed significant, time-dependent reductions in biofilm biomass, reaching 91.2% inhibition after 72 h. This suggests that the nanocomposite interferes with the critical stages of biofilm development, including initial attachment, microcolony formation and maturation.60
The CLSM analysis provided convincing visual and quantitative evidence for the antibiofilm mechanism. Compared to dense, thick (32.5 µm) and predominantly living biofilms on control PS surfaces, biofilms on the nanocomposite were significantly thinner (8.2 µm), sparsely distributed (<20% surface coverage), structurally compromised, and contained a high proportion of dead cells (the ratio of living to dead cells shifted from 4.7:1 to 0.6:1). This dual effect, namely the inhibition of adhesion/growth and killing of embedded bacteria, is highly desirable for an antiplaque material.61, 62 The combination of surface hydrophobicity (reduction of initial adhesion) and sustained AgNP activity (killing of adherent and proliferating bacteria) is presumably the basis for this strong antibiofilm performance.63, 64, 65 The observed efficacy of the material compares favorably with that of other advanced antibiofilm dental materials containing active ingredients such as quaternary ammonium compounds or other nanoparticles.66, 67
The bactericidal mechanism of the PS–HAp–ZrO2–Ag nanocomposite is primarily attributed to the presence of AgNPs, which release Ag+ ions and generate ROS, damaging bacterial cellular components and preventing replication. The hierarchical structure provided by HAp and ZrO2 nanoparticles may also contribute to the overall antimicrobial efficacy by inhibiting bacterial adhesion.
While the developed PS–HAp–ZrO2–Ag nanocomposite demonstrates significant improvements in mechanical strength, antibacterial activity and self-cleaning properties, its practical performance in dental applications must be considered. Key factors include wear resistance, color stability and adhesion to dental tissues. Wear resistance is crucial for restorative materials, as they are subjected to masticatory forces and abrasive food particles.68, 69, 70 The incorporation of ZrO2 nanoparticles is expected to enhance wear resistance due to their high hardness and toughness.71 Color stability is also essential for aesthetic restorations, as discoloration can compromise the material’s appearance.72 The presence of AgNPs may potentially affect color stability due to potential oxidation and subsequent darkening.73 Adhesion to dental tissues is another critical factor, as it directly impacts the material’s ability to bond with tooth structures and withstand clinical stresses.74 The incorporation of HAp nanoparticles may promote biointegration and enhance adhesion to dental tissues due to their structural similarity to natural tooth enamel.68
In this study, HAp, ZrO2 and AgNPs were successfully integrated into a PS matrix, creating a multifunctional nanocomposite with improved mechanical properties, increased hydrophobicity, strong antibacterial activity, and significant antibiofilm efficacy against S. mutans. The novelty of this work lies in the specific combination of these nanoparticles within a polymer matrix to achieve several desired functionalities for potential dental applications such as coatings, temporary restorations or orthodontic components.
Limitations and further directions
Despite the promising results, this study has several limitations that must be acknowledged. Firstly, PS was used as a model polymer matrix due to its low cost, ease of processing and compatibility with nanoparticles. However, PS is not clinically relevant for dental applications, which typically rely on dental-specific polymers such as polymethyl methacrylate (PMMA) or bisphenol-A glycidyl methacrylate (Bis-GMA)-based resins. Therefore, translating this concept to clinically relevant materials would require significant adjustments in nanoparticle dispersion methods, resin compatibility and curing protocols to ensure homogeneous integration and optimal mechanical performance. Secondly, the antibacterial and antiplaque assessments were conducted in vitro using a single bacterial species, S. mutans. While S. mutans is a well-established model for early caries development, natural dental plaque is a complex multispecies biofilm comprising hundreds of bacterial species, including gram-negative anaerobes (e.g., P. gingivalis, F. nucleatum), that play key roles in plaque maturation, immune modulation and periodontal disease progression. Thus, future studies should evaluate the performance of the nanocomposite against multispecies biofilms to better reflect clinical conditions and assess selective antibacterial activity and microbiome balance. Thirdly, the long-term stability, biocompatibility and safety of the nanocomposite require thorough investigation. Although HAp and ZrO2 are generally considered biocompatible, the release kinetics of AgNPs and Ag+ ions remain a concern. Chronic exposure to Ag+ ions may lead to cytotoxicity, microbial resistance or disruption of the oral microbiome. Therefore, comprehensive biocompatibility assessments in accordance with ISO 10993 standards are essential, including in vitro cytotoxicity, genotoxicity and in vivo toxicity studies. Additionally, the optimization of AgNP concentration is crucial to balance antibacterial efficacy with safety. Fourthly, several critical dental properties were not evaluated in this study, including abrasion resistance, color stability and adhesion to dental tissues. These factors are vital for the clinical success of any dental restorative material. For example, color stability ensures aesthetic durability, while strong adhesion to enamel and dentin is essential for marginal integrity and long-term retention.
To address these limitations, future research should focus on the following:
• material translation: developing methods to integrate HAp, ZrO2 and AgNPs into dental resins (e.g., PMMA, Bis-GMA) with improved dispersion and interfacial bonding;
• multispecies biofilm testing: evaluating the antiplaque and antibacterial efficacy against clinically relevant multispecies biofilms, including cariogenic and periodontopathogenic consortia;
• comprehensive biocompatibility assessment: conducting ISO 10993-compliant tests, including cytotoxicity, genotoxicity and in vivo studies to evaluate long-term safety;
• degradation and stability studies: assessing material degradation under simulated oral conditions (thermal cycling, pH cycling, wear) to ensure long-term durability;
• mechanical and functional properties: evaluating abrasion resistance, color stability and adhesion strength to enamel and dentin to ensure clinical feasibility;
• microbiome impact studies: investigating the effects of the nanocomposite on oral microbial ecology, including plaque composition, bacterial diversity and biofilm architecture;
• optimization of formulation: optimizing the nanoparticle loading ratios, dispersion techniques and material composition to enhance performance and minimize risks.
Addressing these aspects would facilitate the translation of the nanocomposite into a next-generation dental material that not only provides structural integrity but also actively promotes oral health through antibacterial, antiplaque and self-cleaning properties.
Conclusions
In summary, this study reports the successful development of a novel PS-based nanocomposite reinforced with HAp, ZrO2 and Ag nanoparticles. The material exhibited significantly improved mechanical strength, increased surface hydrophobicity indicating self-cleaning potential, strong bactericidal activity against S. mutans, and remarkable efficacy in inhibiting biofilm formation. The uniform dispersion of the nanoparticles and their synergistic contributions to the overall performance of the composite were confirmed. Although further optimization as well as extensive biocompatibility and long-term testing are required, especially in clinically relevant polymer matrices, this work provides a strong proof of concept for the development of multifunctional dental materials. Such materials have the potential to actively combat bacterial colonization and plaque formation, thereby contributing improved oral health outcomes.
Ethics approval and consent to participate
Not applicable.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Use of AI and AI-assisted technologies
Not applicable.