Abstract
Background. Oral lichen planus (OLP) is a chronic inflammatory condition affecting the oral mucosa. The oral microbiome has been identified as a potential contributing factor to OLP.
Objectives. The aim of the study was to evaluate the prevalence and diversity of the oral microbiota in patients with OLP.
Material and methods. This observational study included 78 patients with clinically and histopathologically confirmed OLP, recruited in accordance with the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guidelines. Buccal mucosa samples were collected using standardized protocols. DNA was extracted from 12 high-quality samples and subjected to 16S rRNA gene amplification and sequencing. Alpha and beta diversity indices were calculated using the Quantitative Insights Into Microbial Ecology (QIIME) platform. Statistical analyses were performed using the IBM SPSS Statistics for Windows software, v. 26.0 (IBM Corp., Armonk, USA) (p < 0.05).
Results. Intratissue bacterial communities exhibited decreased alpha diversity and increased beta diversity compared with those present on the mucosal surface. Streptococcus, a genus within the Firmicutes phylum, was found to be the most abundant, with 5 Streptococcus strains identified in the OLP samples. Following Streptococci, Bacilli and Clostridia displayed considerable diversity. Other frequently detected species included Klebsiella pneumoniae, Escherichia coli, Pseudomonas aeruginosa, Eikenella corrodens, Actinobacillus, as well as members of the Proteobacteria phylum, which are commonly found in high quantities in the oral cavity. Prevotella and Capnocytophaga, belonging to the Bacteroidetes phylum, were also frequently observed. Alpha diversity analysis revealed significant differences in the colony numbers of the investigated species across studied samples.
Conclusions. The findings indicate an association between the composition of the oral microbiota and OLP. The microbial populations obtained from affected individuals exhibited distinct bacterial compositions. Modulation of the oral microbiome may represent a potential strategy for improving the management of OLP.
Keywords: oral lichen planus, diversity, microbial population, oral microbiota
Introduction
Lichen planus (LP) is an autoimmune condition in which the immune system targets the skin, mucosa, hair, or nails. In some cases, this process may be triggered by external factors such as medication or microbial agents.1 Recent epidemiological data suggests that oral lichen planus (OLP) affects approx. 0.5–2.0% of the global population. The condition typically presents between the ages of 30 and 60, and is more prevalent in women. The development of OLP in children is infrequent.1, 2
Available evidence indicates that immunological mechanisms play a role in the etiology of OLP. Genetic predisposition has also been identified as a potential risk factor, aligning with the STrengthening the REporting of Genetic Association studies (STREGA) guidelines for reporting genetic associations in autoimmune diseases. The disease is more prevalent in women and middle-aged individuals, with an estimated prevalence of 0.1–2.2%. However, the reported prevalence rates and other epidemiological indicators vary considerably across studies.1, 2, 3 In addition to genetic predisposition, several factors such as stress, immunological reactions and trauma contribute to the development of oral lesions associated with OLP.4
Increasing attention has been given to the potential role of the oral microbiome in the development of OLP. Examples of microbes that inhabit the oral cavity include Streptococcus and Corynebacterium species. Together, these microorganisms form an ecosystem that functions in a manner analogous to the human gut microbiota.5, 6 Each region of the oral cavity possesses distinct surface characteristics, giving rise to the presence of different microbial communities. The tongue contains diverse microbial samples when compared to the gums or teeth since their surfaces provide suitable habitats for a variety of microbes.
The oral microbiome comprises over 700 bacterial species, including both aerobic and anaerobic bacteria. Important anaerobic genera include Fusobacterium, Prevotella, Porphyromonas, Treponema, and Tannerella, which play crucial roles in maintaining oral homeostasis but may also contribute to pathological conditions when dysbiosis occurs. Dental plaque and the tongue surface are densely populated with microbes and are considered the most microbially dense habitats.7, 8
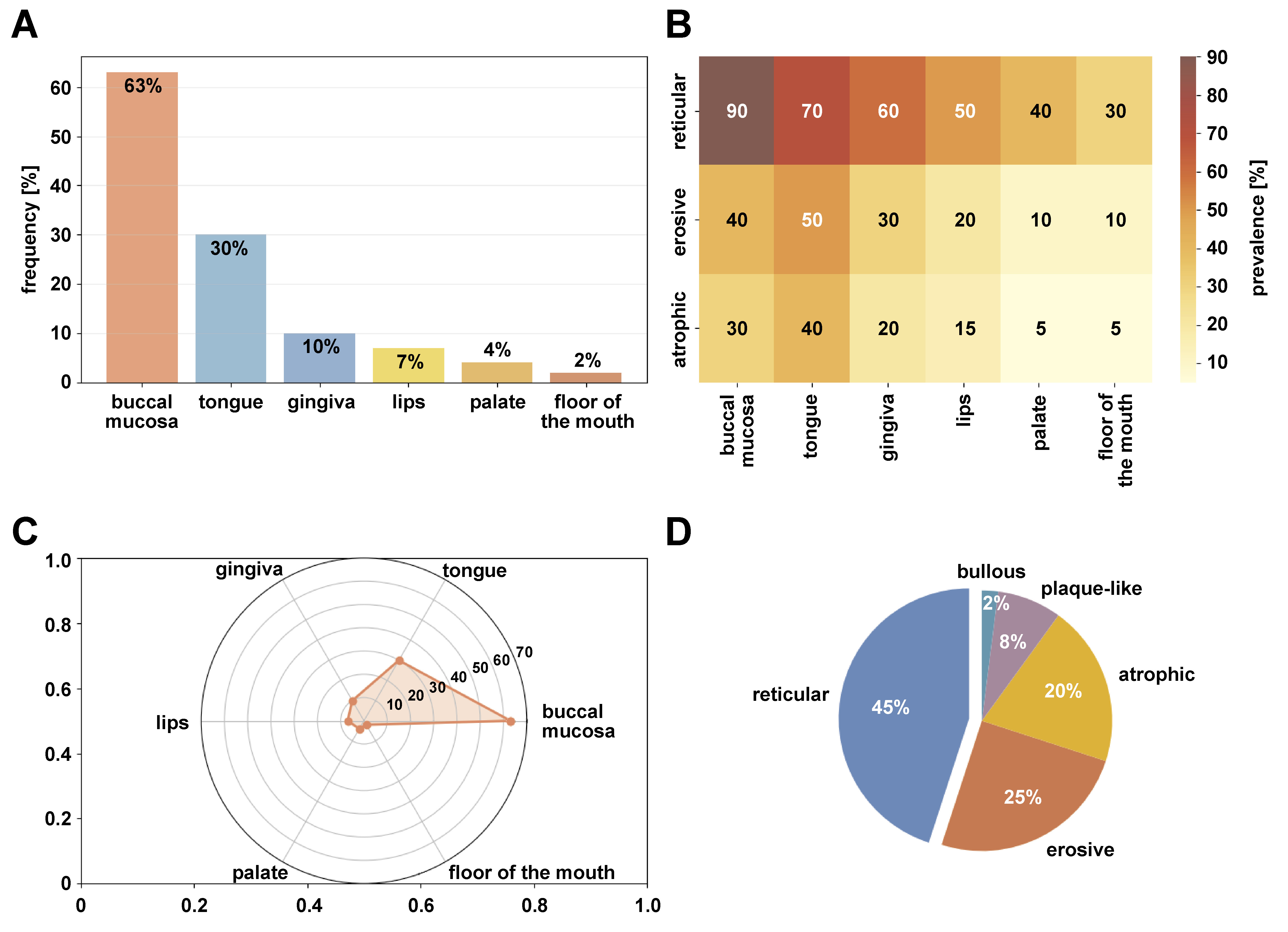
The clinical presentation of OLP varies considerably, with multiple subtypes and anatomical distributions observed in affected patients (Figure 1). The buccal mucosa is the most affected site (63%), followed by the tongue (30%) and gingiva (10%). Lesions typically occur bilaterally and symmetrically. Among the clinical variants, reticular OLP is the most prevalent (45%), characterized by white lacy striae (Wickham’s striae), while erosive (25%) and atrophic (20%) forms represent the symptomatic variants that often require therapeutic intervention (Figure 1).1, 2, 7, 8
Given the important role of the oral microbiome in the occurrence of oral diseases, its regulation is of utmost importance. Studies suggest that the disruption of the homeostatic balance of the oral microbiome leads to the activation of the defense pathways involved in oral inflammation associated with OLP.9, 10, 11
To date, contradictory reports have been published regarding the influence of the oral microbial composition and saliva structure on OLP. Some studies have reported elevated levels of Porphyromonas and Solobacterium in OLP,12 whereas a 2017 report have identified increased abundance of Fusobacterium, Leptotrichia and Lautropia in the buccal mucosa.13 Furthermore, the infiltration of T cells into the oral tissue due to the presence of bacteria in the layers of LP tissue has been reported.14, 15
Studies demonstrate that an individual’s immune status and the level of inflammation influence the diversity and composition of tissue and salivary microbial communities. The structure of an individual’s microbial community may affect the development and progression of OLP.14, 16, 17, 18 Therefore, further research in the fields of immunology and microbiology is necessary to better understand the exact etiology of this disease.
The main objective of the present study is to investigate the bacteria present in the oral cavity of patients with OLP and to explore their potential role in the pathogenesis and development of the disease, which may provide a biological approach to prevent OLP. Specifically, this study aims to: (1) characterize bacterial diversity in OLP lesions using 16S rRNA gene sequencing; (2) identify predominant bacterial taxa associated with OLP; and (3) assess differences in microbial composition between samples.
Material and methods
Study design and setting
This observational study was conducted in accordance with the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guidelines and the STREGA extension for genetic epidemiology studies.
Study population
Consecutive patients presenting with oral mucosal lesions clinically suggestive of OLP were screened for eligibility at the Department of Oral Medicine (Tehran University of Medical Sciences, Iran). The sample size was calculated based on previous microbiome studies in oral diseases, indicating that a minimum of 10 samples is required for 16S rRNA gene sequencing to achieve 80% power at α = 0.05.19
Sample collection and clinical assessment
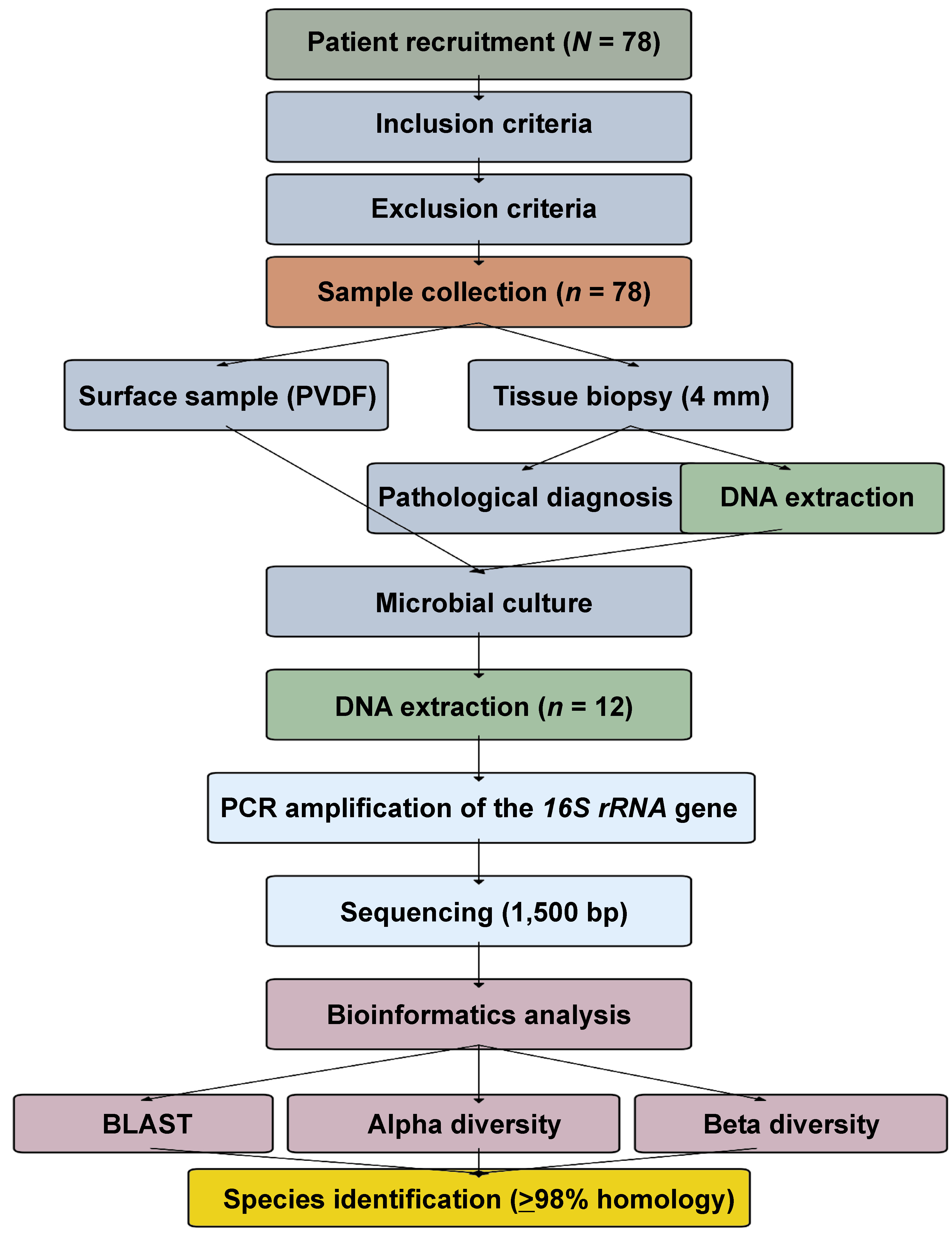
The study was approved by the Institutional Review Boards of the Dental Hospital of Tehran University of Medical Sciences, Iran, and the Faculty of Dentistry, Seoul National University, South Korea (approval No. S-D20180026), as well as by the Ethics Committee of the Clinic of Košice, Slovakia (approval No. REK01/2023). Written informed consent was obtained from all participants. All patients underwent standardized clinical examination performed by 2 calibrated examiners (inter-examiner κ = 0.85). The study workflow is presented in Figure 2. Reticular lesions, without ulceration and with or without erythema, located on the buccal mucosa, were included. Bacteria present on the mucosal surface of the lesion were collected by placing a sterile 20-mm polyvinylidene difluoride (PVDF) membrane on the buccal mucosa for 30 s.
Subsequently, two 4-mm punch biopsy specimens were obtained: one was used to create a tissue block for pathological diagnosis, while the other was utlilized to extract DNA for microbiota analysis.
Inclusion and exclusion criteria
The inclusion criteria were as follows: clinically and histopathologically confirmed diagnosis of OLP; willingness to participate in the study; no history of antibiotic or steroid treatment in the past month; and absence of infectious diseases.
The exclusion criteria included the withdrawal of consent, low unstimulated whole salivary flow rate (≤0.1 mL/min), and smoking. Unstimulated whole saliva was collected at baseline over a 5-min period to determine salivary flow rate. All included subjects exhibited salivary flow rates that exceeded the exclusion threshold (range: 0.21–0.49 mL/min).
Screening and quantification of microorganisms in samples
Serial dilutions (1−10 to 10−10) were prepared to assess the number of microorganisms present in the OLP samples. Under standard and sterile conditions, 0.5 g of each sample was transferred to the tube with sterile distilled water. The contents of the first tubes were mixed until the dilution was completely uniform. Then, 1 mL of the suspension was transferred into subsequent tubes under sterile conditions. One milliliter of each dilution was transferred to the agar culture medium under standard and sterile conditions and spread uniformly using a sterile pipette. Two plates were considered for each dilution. The plates were incubated at 25°C for 7 days. The average number of countable colonies was obtained, and the number of culturable bacteria per gram of sample was calculated based on dilution factors. Single colonies formed on the culture media were linearly cultivated to ensure the purity of oral bacteria.
DNA extraction
A total of 12 samples were selected for DNA extraction based on the predefined quality criteria: absence of blood contamination; sufficient tissue yield (>40 mg); and intact morphology. A DNA extraction kit (MACHEREY-NAGEL GmbH & Co. KG, Düren, Germany) was used to extract DNA from the buccal mucosa and tissue samples following the manufacturer’s protocol, with modifications for oral tissue samples.
The following steps were performed during the DNA extraction process. The cells were collected from a culture medium by centrifugation in a microtube. A sample of 40 mg wet weight of the microbial cell culture medium plate was used. Finally, 100 µL of BE buffer solution was added, and a cell suspension was prepared. To homogenize and lyse the samples, the cell suspension was transferred to the NucleoSpin Bead Tube Type B (MACHEREY-NAGEL GmbH & Co. KG). The glass beads were subjected to a process of regeneration to ensure the removal of any remaining cell debris at the end of the tube. After centrifugation, 500–600 µL of the NucleoSpin Bead Tube column supernatant was transferred to a 2-mL tube. Subsequently, 500 µL of buffer B was added and centrifuged for 30 s at 11,000 g, and the column solution was collected in tubes. Then, 500 µL of B5 buffer was added to the column, centrifuged for 30 s at 11,000 g, and the column was collected in tubes. To dry the membrane, the column was centrifuged for 30 s at 11,000 g, removing any remaining washing buffer. The Nucleospin Bead Tube column was transferred to a 1.5-mL tube, and 100 µL of BE buffer was added to the column that was then incubated for 1 min at room temperature. Subsequently, the sample was centrifuged at 11,000 g for 30 s. The presence and quantity of DNA were evaluated by electrophoresis on 1% agarose gel. Additionally, the concentration of extracted DNA was measured using a spectrophotometer (NanoDrop Spectrophotometer; Thermo Fisher Scientific, Waltham, USA).
Polymerase chain reaction
Polymerase chain reaction (PCR) is a widely used technique in molecular biology and genetic disease research for the identification of new genes. The activity of genes can be measured through the implementation of PCR on their RNA. The conditions for performing PCR were as follows: a 2X PCR Master Mix kit was obtained from SinaClon BioScience Co. (Tehran, Iran). Selective primers for the amplification of the 16S rRNA gene, designated as forward 9 and reverse 1541, were procured from Elgo Fanavaran Pars DNA Co. (Tehran, Iran), and their complete specifications are listed in Table 1.
After conducting PCR in accordance with standard protocols, the integrity of the amplicons was assessed, revealing a consistent band size of 1,500 base pairs. Following electrophoresis, purification was executed by TopazGene (Kamal Shahr, Iran) and Microsens (Lausanne, Switzerland), bypassing the cloning stage. Subsequently, sequencing was undertaken, and the resulting sequences were analyzed using the Basic Local Alignment Search Tool (BLAST). For agarose gel preparation, the volume of the cassette was instrumental in determining the required quantities of agarose gel powder and TBE buffer, ensuring a suitable matrix for electrophoretic separation.
Statistical analysis
During the statistical analysis, sequence identifications were aligned against the National Center for Biotechnology Information (NCBI) gene database. Strains exhibiting ≥98% sequence homology were grouped collectively. Alpha diversity metrics, including Chao1, abundance-based coverage estimator (ACE), and Shannon and Simpson indices, quantified species complexity, which was analyzed using the Quantitative Insights Into Microbial Ecology (QIIME) v. 2.1.0 platform (https://qiime2.org). Beta diversity assessments were conducted using QIIME v. 1.7.0. Statistical analyses were performed using the IBM SPSS Statistics for Windows software, v. 26.0 (IBM Corp., Armonk, USA). Differences between groups were evaluated using one-way analysis of variance (ANOVA) for normally distributed data or the Kruskal–Wallis test for non-normally distributed data. Normality was assessed using the Shapiro–Wilk test. A p-value <0.05 was considered statistically significant.
Bias control
To minimize selection bias, consecutive sampling was employed. Laboratory personnel performing molecular analyses were blinded to clinical data. Technical replicates were conducted for 20% of samples to assess reproducibility.
Results
Seventy-eight individuals with clinically and histopathologically confirmed OLP were included in the study. All participants completed the study protocol. The mean age of the patients was 49.70 ±15.54 years (range: 12–71 years), with 61% of the patients being male and 39% being female. Histopathological examination of all cases confirmed the diagnosis of OLP. Among the 78 collected samples, 53 (67.9%) yielded DNA of sufficient quality for initial screening. Of these, 12 samples (15.4%) met all predefined quality criteria for 16S rRNA gene sequencing: DNA concentration >50 ng/µL; A260/A280 ratio of 1.8–2.0; and the absence of PCR inhibitors. Among the buccal mucosa and tissue samples, 53 specimens were cultured, as they were of good quality. Twelve samples from the target cultures were subjected to microbiological analysis (Table 2).
Detection of bacterial DNA in tissue samples
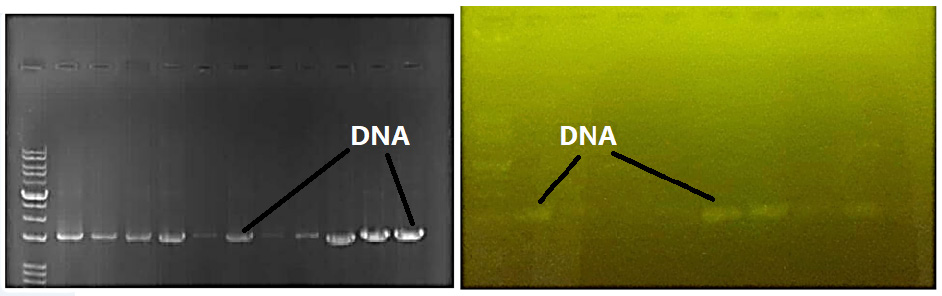
Agarose gel electrophoresis confirmed the detection of bacterial DNA within the samples. Specifically, the validation of the DNA extraction process, yielding bands representative of bacterial genomic material sourced from the buccal mucosa and tissue specimens, is visually presented in Figure 3, attesting to the successful procurement and separation of nucleic acid fragments characteristic of the oral microbiome.
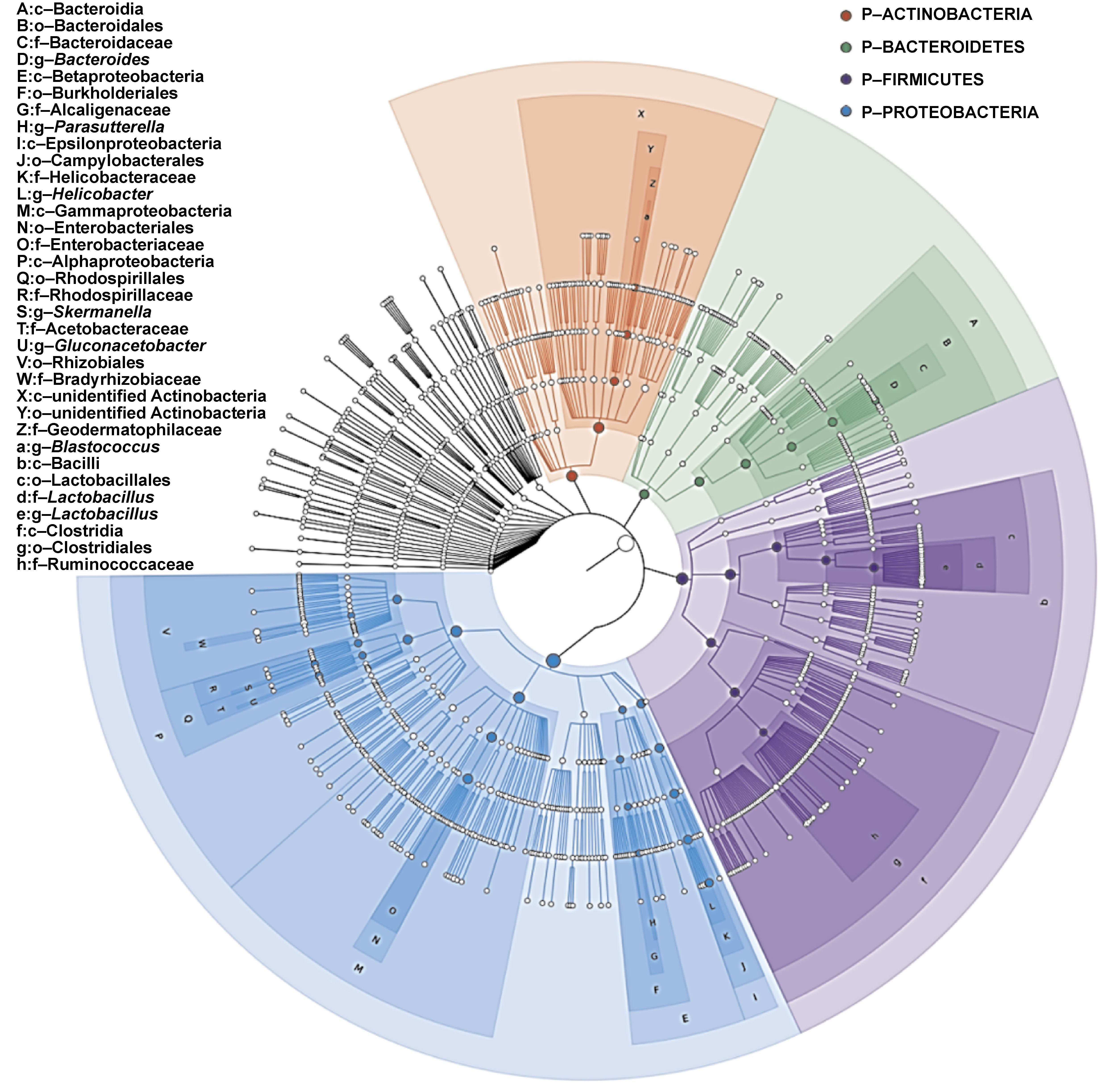
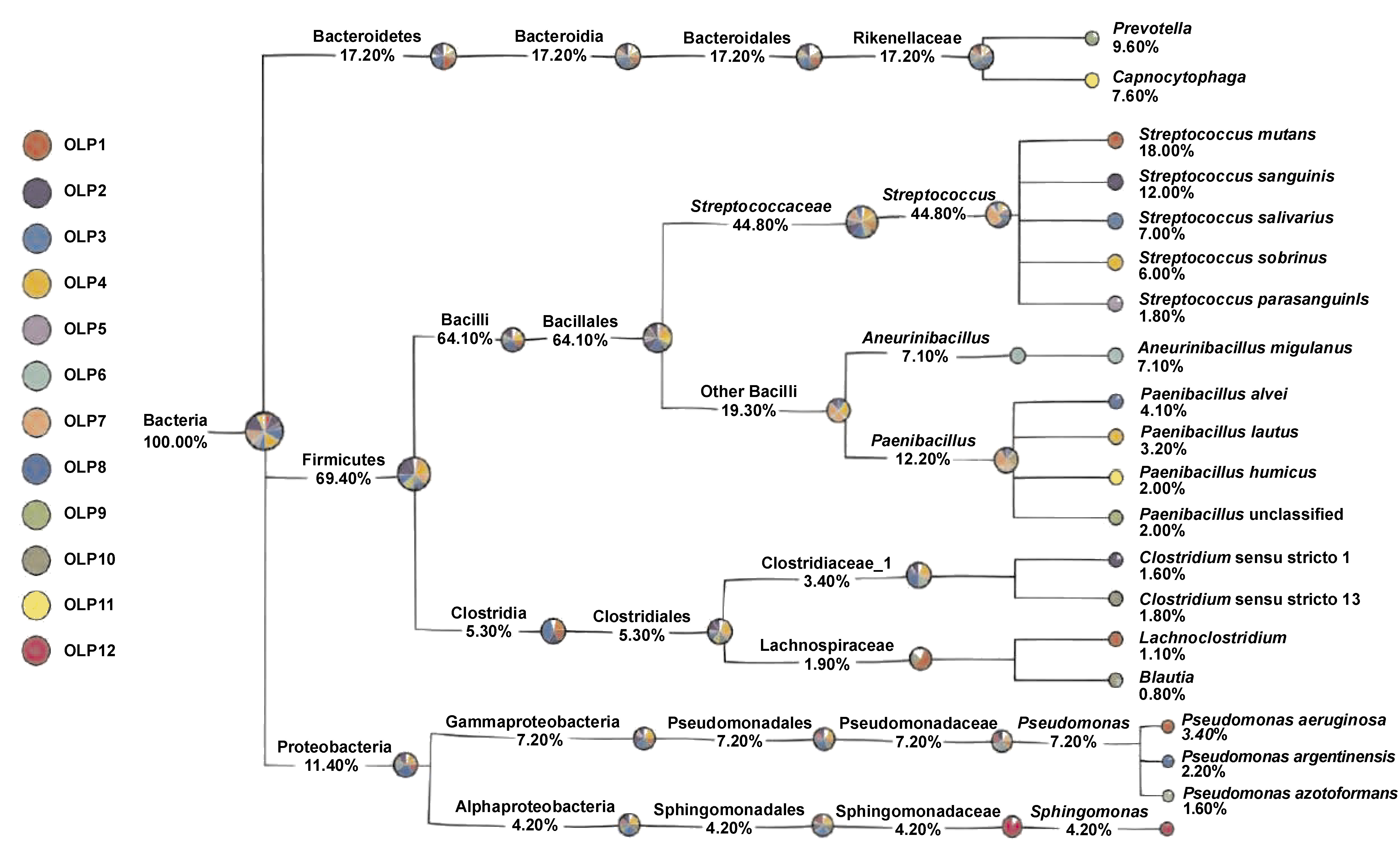
GraPhlAn display analysis
A classification tree was generated for all samples using the GraPhlAn software (Figure 4). The analysis demonstrated that the Firmicutes phylum exhibited a predominant share in the microbial community, with a notable richness in species diversity, and was surpassed only by the Proteobacteria phylum. Within Firmicutes, the genus Streptococcus was the most abundant. The spectrum of the identified species included, but was not limited to, Klebsiella pneumoniae, Escherichia coli, Pseudomonas aeruginosa, Eikenella corrodens, Actinobacillus, and other members of the Proteobacteria phylum, which are well-documented as abundant inhabitants of the oral cavity. Additionally, the Bacteroidetes phylum was represented by genera Prevotella and Capnocytophaga, further enriching the oral microbiota profile.
Gene tree of tissue samples
An evolutionary phylogenetic tree was constructed to represent the species most frequently detected in the sampled cohort, leveraging next-generation sequencing data and the analytical prowess of the R&D software suite of BLAST. As illustrated in Figure 5, the pre-eminence of the Firmicutes phylum was pronounced, with Streptococcus identified as the most prevalent genus. Within this genus, species such as Streptococcus mutans, Streptococcus sanguinis, Streptococcus salivarius, and Streptococcus sobrinus were detected. Following Streptococcus, members of the classes Bacilli and Clostridia exhibited notable phylogenetic breadth. Beyond Firmicutes, Proteobacteria emerged as a significant phylum, comprising species such as E. coli, P. aeruginosa and E. corrodens, as well as Actinobacillus. Collectively, these taxa represent pivotal members within the Proteobacteria phylum, as determined by the study findings.
Distribution of bacterial taxa
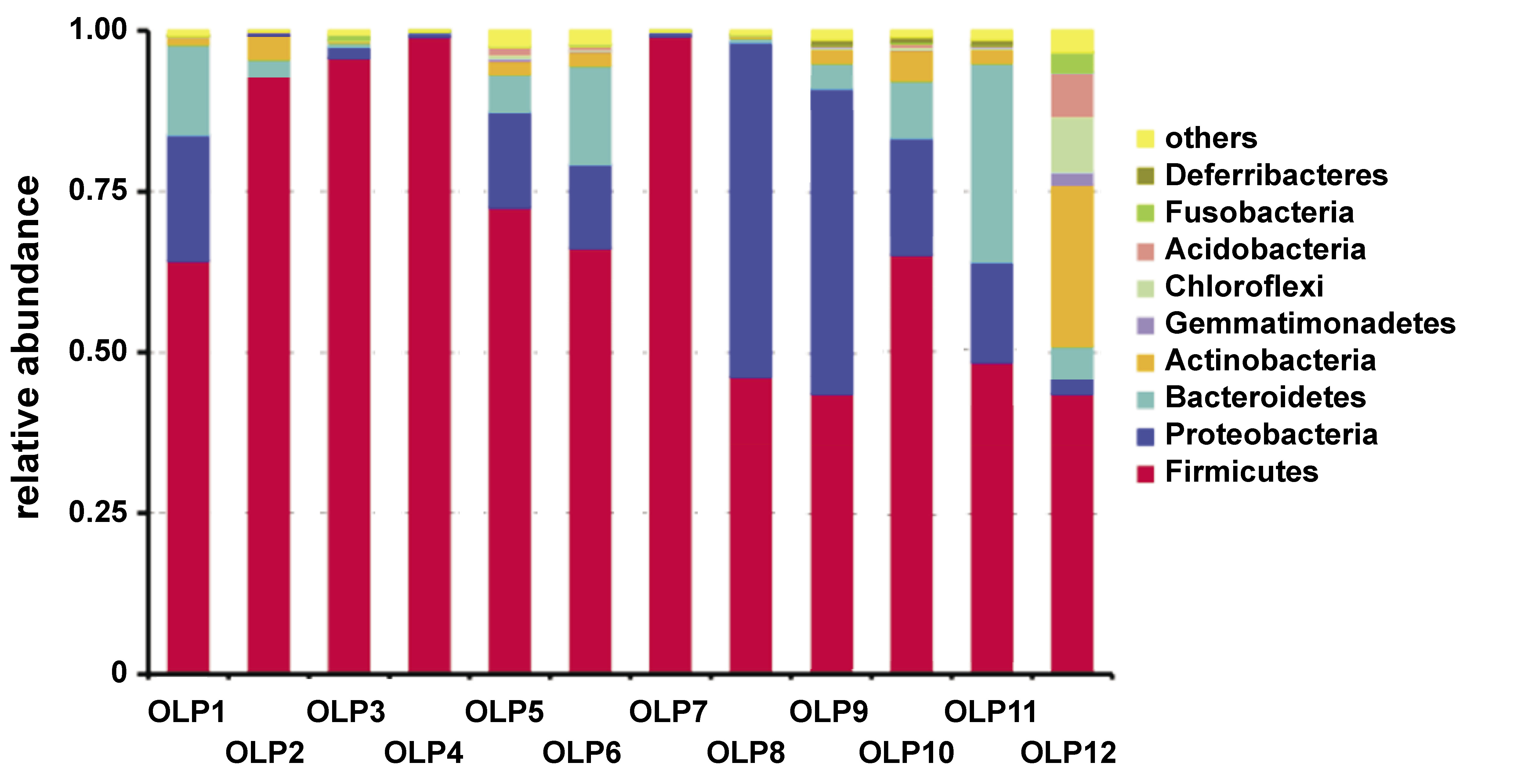
The analysis of bacterial distribution revealed marked disparities in bacterial populations among distinct buccal mucosa and tissue specimens. The genus abundance plot illustrated in Figure 6 demonstrates that Firmicutes constitute the dominant phylum. Notably, samples OLP8, OLP9 and OLP10 exhibited a considerable representation of Proteobacteria. Conversely, samples OLP2, OLP3, OLP4, and OLP12 were dominated by Firmicutes, with comparatively lower proportions of Proteobacteria. Sample OLP12 displayed the highest compositional diversity, characterized by the coexistence of Firmicutes and Actinobacteria.
Sequencing-assisted alpha diversity analysis
Alpha diversity analysis, as initially characterized by Whittaker in 1972, fundamentally quantifies species richness, addressing species variety within a given sample. Empirically, this is achieved by enumerating observable species or operational taxonomic units (OTUs) per sample. The assessment of alpha diversity revealed substantial disparities in the number of colonies corresponding to the investigated species across samples. Indices such as phylogenetic diversity, ACE and Chao1 collectively indicated that samples OLP3 and OLP12 had the highest microbial diversity, whereas OLP2, OLP4, OLP7, and OLP8 exhibited the lowest diversity.
The Shannon index, a measure encompassing both species richness and evenness, demonstrated that OLP3, OLP6, OLP10, and OLP12 had more diverse and evenly distributed microbial communities compared to the remaining samples, whereas OLP7 showed the lowest diversity. The Simpson index, which emphasizes the dominance of certain species, indicated higher diversity in samples OLP3, OLP6, OLP11, and OLP12, with OLP7 again demonstrating the lowest diversity and reflecting a less complex community structure (Table 3). These findings collectively underscore the heterogeneity in the microbial composition among the samples studied.
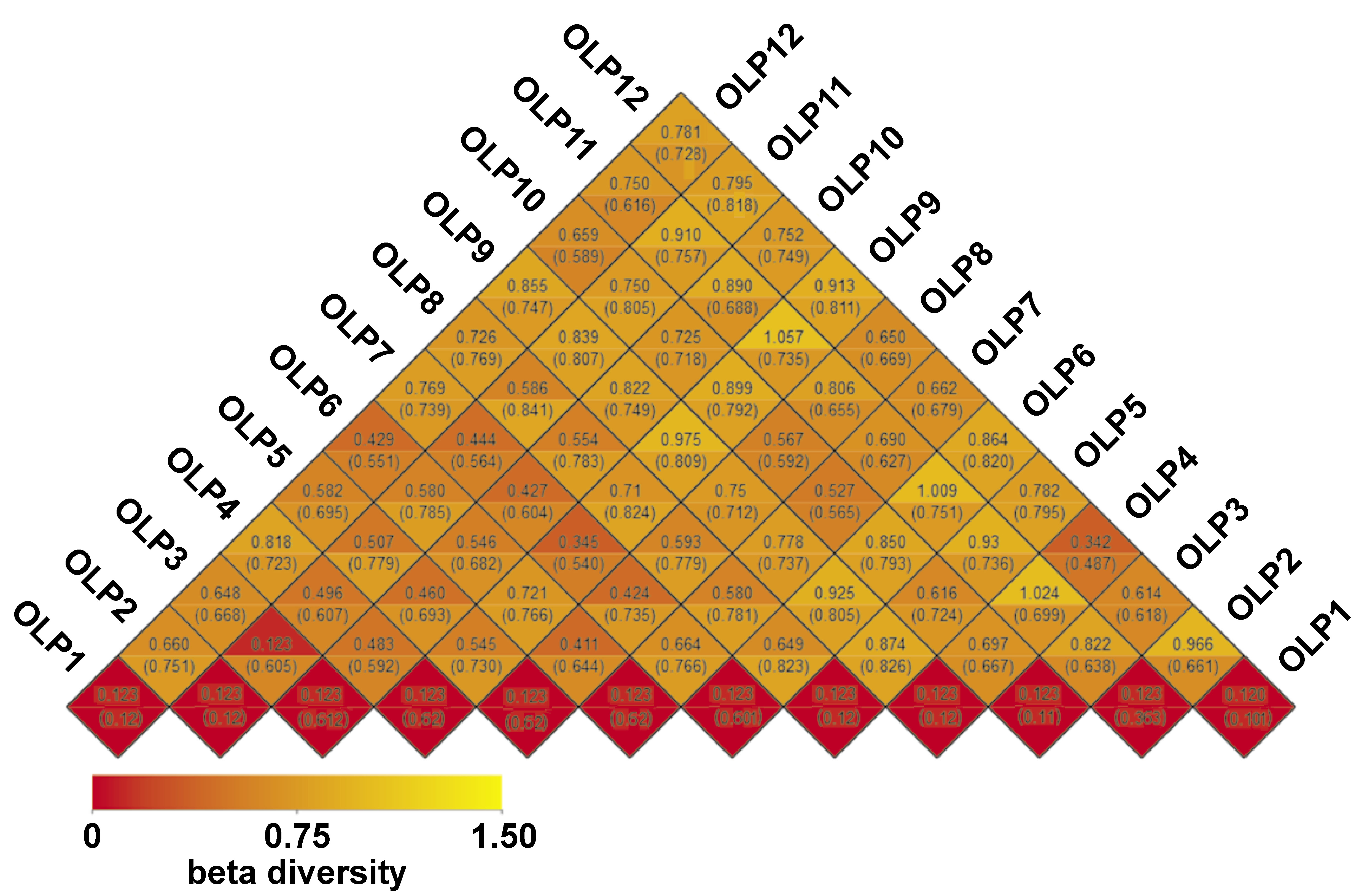
Beta diversity analysis
Beta diversity analysis is a quantitative measure that explicates the distinction in microbial community structures by comparing their compositions. The heatmap analysis indicated that samples OLP12, OLP1, OLP4, OLP8, and OLP3 exhibited the greatest degree of dissimilarity in their microbial compositions. Notably, sample OLP1 demonstrated the highest level of dissimilarity, suggesting a unique microbial profile within the dataset (Figure 7).
Discussion
The present study aimed to identify bacterial species associated with OLP by analyzing intratissue microbiota within OLP lesions. Intratissue bacterial communities were characterized by decreased alpha diversity and increased beta diversity compared with those present on the mucosal surface. The GraPhlAn display analysis revealed that the Firmicutes phylum was the dominant component of the microbial community, followed by the Proteobacteria phylum. Bacteria within the large genus Streptococcus, categorized under the Firmicutes phylum, were the most abundant. Five Streptococcus strains from the OLP samples were confirmed by means of whole genome sequencing. Following Streptococci, Bacilli and Clostridia demonstrated increased diversity. Other identified species included K. pneumoniae, E. coli, P. aeruginosa, E. corrodens, Actinobacillus, and other members of the Proteobacteria phylum. According to previous studies, these species are found in significant quantities within the oral cavity. Prevotella and Capnocytophaga, belonging to the Bacteroidetes phylum, were identified as other prevalent bacteria in the samples. Studies have shown that the composition of saliva and buccal mucosa differs in healthy individuals, with genera such as Neisseria and Prevotella comprising nearly half of the bacterial composition of saliva. The bacterial community observed in OLP patients appears to be altered, with marked differences in diversity. Several reports have identified Neisseria and Prevotella among the predominant genera in OLP.11, 19 Colonization occurs at birth or shortly thereafter, with S. salivarius being one of the pioneer species. During the initial year of colonization, the oral cavity is also subject to invasion by other microorganisms, including Streptococcus, Lactobacillus, Actinomyces, Neisseria, and Veillonella.20 Despite the presence of various microorganisms in the oral cavity, there is a symbiotic relationship between them, with non-pathogenic species generally preventing pathogenic species from adhering to the mucosa. In order to become pathogenic, bacteria must cross the commensal barrier, thus inducing infection and disease.21, 22
The gene tree analysis of the studied samples showed that the members of the Firmicutes phylum, particularly Bacilli, were the most common bacterial species present, followed by Clostridia, S. mutans, S. sanguinis, S. salivarius, and S. sobrinus.
The evaluation of species distribution across samples revealed that in most of the infected patients, the dominant bacterial species belonged to the Firmicutes phylum. However, in samples OLP8, OLP9 and OLP10, members of the Proteobacteria phylum constituted a significant percentage of the bacterial population. In contrast, samples OLP2, OLP3, OLP4, and OLP12 were predominantly composed of Firmicutes, with Proteobacteria present in lower abundance. Sample OLP12 exhibited the highest microbial diversity, including representatives of both Firmicutes and Actinobacteria, whereas OLP8 showed a relatively high abundance of Proteobacteria.
Microbiological studies have reported the presence of various microbes in the oral cavity, including Streptococcus and Corynebacterium species, which collectively form an ecosystem that functions similarly to the human gut microbiota.5, 6 Various factors, including different treatments, can affect the population of oral bacteria.23, 24 The microbial population is influenced by the type and severity of the disease. The structure of oral biofilm is associated with the development of various oral diseases.25, 26, 27 Systemic conditions, such as cardiovascular, digestive or endocrine disorders, may alter the oral microbiota. Body weight may also have an influence, with obese patients having a different salivary microbiome compared to those with normal weight. Additionally, liver diseases significantly alter the salivary microbiota and may invade the gut.28, 29, 30 Sugar consumption, which changes the acidity of the oral environment, leads to modifications in the oral microbiota, with an increase in acidogenic bacteria.31 Diet is a crucial factor in shaping the oral microbiome. For example, breastfed infants tend to have higher proportions of Streptococcus, while formula-fed infants exhibit increased levels of Actinomyces and Prevotella. Moreover, breastfed and formula-fed infants develop oral candidiasis less frequently than solid-fed infants.32, 33, 34 Smoking has been shown to alter the oral flora by reducing Proteobacteria and increasing Firmicutes and Actinobacteria.35, 36
Notably, in the present study, alpha diversity analysis revealed substantial inter-individual variability. Sample OLP7 exhibited the lowest microbial diversity (Shannon index: 0.78, Simpson index: 0.18), whereas OLP12 demonstrated the highest diversity (Shannon index: 8.16, Simpson index: 0.98). Clinically, OLP7 presented with higher erythema (R5E5) and a moderate reticulation/erythema/ulceration (REU) score, while OLP12 demonstrated lower erythema (R6E2) but a higher REU score. Histopathological examination confirmed the diagnosis of OLP in both cases without secondary infection or atypical dysplasia. As no clear differences in comorbidities, systemic diseases or medication use were identified between these 2 subjects, the observed variation in microbial diversity may reflect individual host–microbiome interactions or localized microenvironmental factors. This observation highlights the potential significance of patient-specific microbial diversity profiles in the pathogenesis of OLP.
Numerous studies have investigated various aspects of the etiology and treatment of OLP. Changes in saliva and its components may lead to a wide range of oral disorders and diseases.37 Two types of peroxidase enzymes, which function as crucial defense factors, are secreted by salivary glands and polymorphonuclear leukocytes. The latter is discharged in the gingival crevice fluid. Peroxidase enzymes, in the presence of thiocyanate and hydrogen peroxide ions, cause the destruction of Lactobacillus acidophilus. They play a crucial role in fighting microbes and preventing the accumulation of harmful amounts of hydrogen peroxide.38 In OLP, disturbances in redox homeostasis have been reported, including reduced antioxidant capacity and increased oxidative stress. Elevated levels of lactoferrin, lysozyme and salivary peroxidase have been observed in bacterial or viral infections. Increased levels of peroxidase enzymes in patients may contribute to oxidative imbalance, which could potentially influence autoimmune responses.37, 38
The role of oxidative stress in oral inflammatory diseases has been extensively studied. Recent investigations have shown significant alterations in both enzymatic and non-enzymatic antioxidant systems in periodontitis. Toczewska et al. demonstrated reduced activity of antioxidant enzymes and decreased levels of non-enzymatic antioxidants in saliva and gingival crevicular fluid of patients with periodontitis.39, 40 These findings parallel our observations in OLP, suggesting shared oxidative stress pathways in chronic oral inflammation. Moreover, the complexity of diagnosing and managing oral mucosal inflammatory conditions, as highlighted in the review of peri-implant mucositis by Lo Bianco et al.,41 emphasizes the need for standardized diagnostic criteria and therapeutic protocols.
In the present study, the findings of alpha diversity analysis showed that the number of colonies associated with the investigated species exhibited significant differences and diversity among samples. The phylogenetic diversity index, as well as the ACE and Chao1 indices, revealed that OLP3 and OLP12 had higher microbial diversity, while the lowest diversity was observed in OLP2, OLP4, OLP7, and OLP8 samples. The Shannon index indicated that OLP3, OLP6, OLP10, and OLP12 demonstrated a greater number of microbial species than other samples. The Simpson index showed that the dominant species in OLP3, OLP6, OLP11, and OLP12 exhibited more microbial species than the other samples. These findings align with previous reports suggesting that increased disease severity may be associated with reduced microbial diversity.13, 42, 43
The findings of our study also demonstrate that E. coli, an indicator of oral contamination, was identified in abundance in patients with OLP. After Streptococcus, Escherichia coli has been identified as an effective pathogen in OLP. A recent study isolated 4 Streptococcus strains from additional OLP samples, and detected E. coli in most of the OLP tissues, suggesting its potential role in the pathogenesis of OLP.44
Contradictory reports have been published regarding the effect of the oral microbial community structure and saliva on OLP. Some reports indicate the presence of high levels of Porphyromonas and Solobacterium in OLP, while a 2017 report found that Fusobacterium, Leptotrichia and Lautropia exhibited high abundance in the buccal mucosa.13 Additionally, the infiltration of T cells into oral tissues due to the presence of bacteria in OLP has been reported.14, 15
Studies have shown that when the homeostatic balance of the oral microbiome is disrupted, the defense pathways involved in oral inflammation associated with OLP are activated. In other words, the occurrence of OLP is associated with the disruption of the oral microbiota due to microbial dysbiosis of the buccal mucosa.9, 10, 11
Study strengths and limitations
This study has several strengths, including the use of standardized clinical criteria, histopathological confirmation of all cases, and high-throughput 16S rRNA gene sequencing for comprehensive microbial profiling. The application of both alpha and beta diversity measures enabled a thorough assessment of microbial community structure.
However, several limitations should be acknowledged. First, the observational design precluded causal inference regarding the relationship between microbial dysbiosis and the development of OLP. Second, the absence of a matched control group limited direct comparisons. However, the findings were compared with published data on healthy oral microbiota. Third, the final sample size for sequencing (n = 12) was relatively small due to stringent quality criteria, potentially limiting statistical power for subgroup analyses. Fourth, functional aspects of the microbiome or host–microbe interactions were not assessed. Finally, potential confounding factors such as diet, oral hygiene practices and subclinical systemic conditions were not systematically evaluated. Future longitudinal studies with larger sample sizes, matched controls and functional microbiome analyses are warranted to confirm these findings and elucidate the mechanistic role of the oral microbiome in OLP pathogenesis.
Conclusions
The present study demonstrated an association between the oral microbiome and OLP. The microbes obtained from samples of OLP patients exhibited a distinct bacterial population. Overall, the predominant bacterial genera were Streptococcus, followed by Bacillus. The difference in bacterial population may serve as an influential factor in the development of OLP. It is recommended that the bacteria identified in this study be compared with healthy oral microbiota and further evaluated in order to enhance understanding of the pathogenesis of OLP. While the present study identified facultative anaerobes, such as Streptococcus species, the potential role of obligate anaerobic bacteria in the pathogenesis of OLP warrants further investigation, particularly given their abundance in oral biofilms and their capacity to modulate local immune responses.
Ethics approval and consent to participate
The study was approved by the Institutional Review Boards of the Dental Hospital of Tehran University of Medical Sciences, Iran, and the Faculty of Dentistry, Seoul National University, South Korea (approval No. S-D20180026), as well as by the Ethics Committee of the Clinic of Košice, Slovakia (approval No. REK01/2023). Written informed consent was obtained from all participants.
Data availability
All data generated or analyzed during this study are included in the published article.
Consent for publication
Not applicable.
Use of AI and AI-assisted technologies
Not applicable.