Abstract
In recent years, significant advancements in the understanding of the processes underlying heart failure (HF) have been made, particularly regarding the role of chronic low-intensity inflammation or smoldering inflammation (SI). This review consolidates findings from the available literature and illustrates the relationships between inflammation, neurohormonal activation, metabolic derangements, and comorbidities in HF, with a focus on heart failure with preserved ejection fraction (HFpEF).
A comprehensive literature search was conducted using PubMed®, Wiley Online Library, Scopus, and Web of Science (limited to 2025). The search terms included “heart failure”, “HFpEF”, “inflammation”, “smoldering inflammation”, “biomarkers”, “cytokines”, “fibrosis”, and “comorbidities”. Peer-reviewed articles, reviews, as well as clinical and observational studies describing the mechanistic, prognostic and therapeutic aspects of SI in HF were included. Studies limited to acute coronary syndrome (ACS) were excluded.
Structural changes leading to hemodynamic perturbations in HFpEF are correlated with processes mediated by SI. Several biomarkers measure inflammation and provide diagnostic and prognostic value, including C-reactive protein (CRP), interleukin-6 (IL-6), soluble suppression of tumorigenicity 2 (sST2), galectin-3 (Gal-3), and iron homeostasis. Clinical trials demonstrate the efficacy of sodium–glucose cotransporter-2 (SGLT-2) inhibitors, glucagon-like peptide 1 (GLP-1) receptor agonists, and other targeted interventions in the modulation of SI.
Smoldering inflammation is a key mechanism in the pathogenesis of HFpEF and the progression of comorbidities. Understanding SI may improve risk stratification and management strategies. Both established and emerging anti-inflammatory therapies, when administered alone or in combination, may target SI in order to enhance HF management.
Keywords: inflammation, heart failure, cardiac remodeling, neurohormonal interactions
Introduction
Heart failure (HF) is a cardiac condition that has emerged as a global health concern, affecting approx. 64 million individuals worldwide.1 Recent advancements in pharmacotherapy for one of the HF phenotypes, HF with reduced ejection fraction (HFrEF), have shifted the structure of patients presenting with a specific HF phenotype toward HFpEF. Nowadays, HFpEF accounts for more than half of the patients presenting with HF.2 Due to its heterogeneous etiology and complexity of development, managing this condition is still a significant challenge. One of the potential factors contributing to the pathophysiology of HF, and especially to HFpEF, is smoldering inflammation (SI), which can be described as chronic low-grade inflammation, promoting maladaptive responses and triggering maladaptive mechanisms.3
Inflammation is associated with neurohormonal activation, fibroblast activation, and subsequent fibrosis, oxidative stress, vascular endothelial dysfunction, and ischemia.3 These processes are the fundamental core of SI. The chronic nature of this condition complicates its observation and analysis. However, in recent decades, the complex relationship between metabolic pathways and the onset and development of HF has been revealed.4 Consequently, we have identified specific inflammation-related biomarkers that might assist in predicting the future onset of HF that remains clinically silent.5 Additionally, inflammatory indicators may prove useful in risk stratification and outcome assessment.2, 3, 5, 6, 7, 8, 9, 10, 11, 12
Comorbidities that are prevalent in patients with HF should be perceived not only as additional conditions, but rather as a common result of a shared pathophysiological, inflammation-dependent process or even a direct cause of HF development.13, 14, 15, 16, 17, 18, 19, 20, 21, 22 It is important to distinguish the connection between HF and comorbidities because effective treatment of concomitant diseases is a promising way of preventing HF onset or progression. In the subset of HFpEF patients, SI appears to play a pivotal role in the pathogenesis process, simultaneously promoting the development of multimorbidity and exacerbating its severity.5, 23, 24 Therefore, the role of SI in the etiology of the disease requires further evaluation.
Lastly, pioneering discoveries related to the role of inflammation in the exacerbation of HF provided a rationale for the evaluation of novel drugs for SI alleviation, as well as for the identification of effective responses to the growing number of patients presenting with HFpEF who require advanced treatment.16, 25, 26, 27 Numerous therapeutic avenues related to SI research offer optimism for the identification of efficacious, life-extending therapeutic modalities, but the results of these studies require careful analysis.
Even though numerous studies and reviews have explored the role of inflammation in cardiovascular disease (CVD) and HF, the majority of these studies have focused on either HFpEF or responses to acute inflammation. Smoldering inflammation has not been explored as a mechanism that connects comorbidities, metabolic dysfunction and structural cardiac remodeling in the context of HFpEF. Importantly, none of the reviews has systematically linked biomarkers, comorbidities or therapies into a single construct. Current studies describe biomarkers, comorbidities or therapies separately, and do not provide a comprehensive framework of how chronic, low-grade inflammation contributes to the presentation, progression and therapeutic response of HFpEF. By using available evidence regarding molecular pathways, biomarkers, comorbidities, and novel anti-inflammatory interventions, our objective is to identify and address this knowledge gap by offering a comprehensive view of SI in the context of HFpEF. We hope that our unique approach will compel others to emulate this model in clinical research and alternative therapeutic approaches.
Therefore, the aim of this review was to consolidate the current evidence regarding the role of SI in HF, with a particular focus on HFpEF. Particular attention has been placed on biomarkers, outcomes, comorbidities, and novel therapies to elucidate the associations between SI and the development, exacerbation and treatment of HFpEF.
Material and methods
In this review, a structured literature search was performed for studies assessing the role of SI in the field of HF, with a focus on HFpEF. The core databases used for the search were PubMed® and Scopus, with supplementary searches conducted in Web of Science and Wiley Online Library, if appropriate. The following Medical Subject Headings (MeSH) terms were used: “heart failure”, “HFpEF”, “inflammation”, “smoldering inflammation”, “biomarkers”, “cytokines”, “fibrosis”, and “comorbidities”. These keywords were utilized to assess studies evaluating a range of pathophysiological mechanisms, as well as diagnostic and prognostic aspects of molecular pathways for SI and therapeutic measures implemented in HF.
The search encompassed articles published until 2025, with no other limitations regarding study design. To assess the multifactorial source of inflammation in HF, the study analyzed clinical trials, systematic reviews and observational cohort studies.
The inclusion criteria for the study encompassed peer-reviewed original articles, reviews, meta-analyses, and clinical trials that discussed inflammation, inflammatory biomarkers and inflammation-targeted therapies within the context of HF, with a particular focus on HFpEF. The exclusion criteria were studies that focused solely on acute coronary syndrome (ACS) or non-inflammatory origin of HF. The selection of articles was based on relevance, scientific quality and novelty.
Pathophysiology
The pathophysiology of HF is complex, with inflammation playing a pivotal role in this process. Nevertheless, the contribution of inflammation varies depending on the HF phenotype, with myocardial injury being the leading cause of HFrEF and inflammation in HFpEF.3 Notably, these phenotypes are not distinct entities, with heart failure with mildly reduced ejection fraction (HFmrEF) serving as a buffer between them that can progress to either HFpEF or HFrEF.4, 28 The phenotypes of HF share some common pathways, some of which are more pronounced in HFpEF compared to HFrEF and vice versa.3
Heart failure with preserved ejection fraction is mainly preceded by metabolic diseases, including obesity and type 2 diabetes mellitus (T2DM). These conditions induce chronic low-grade inflammation, also referred to as metainflammation.29 Metainflammation leads to coronary microvascular endothelial inflammation, which contributes to cardiac remodeling through fibrosis and hypertrophy.30, 31 The aforementioned mechanisms result in left ventricular (LV) stiffness and increased filling pressure at rest and during exercise.32, 33, 34, 35
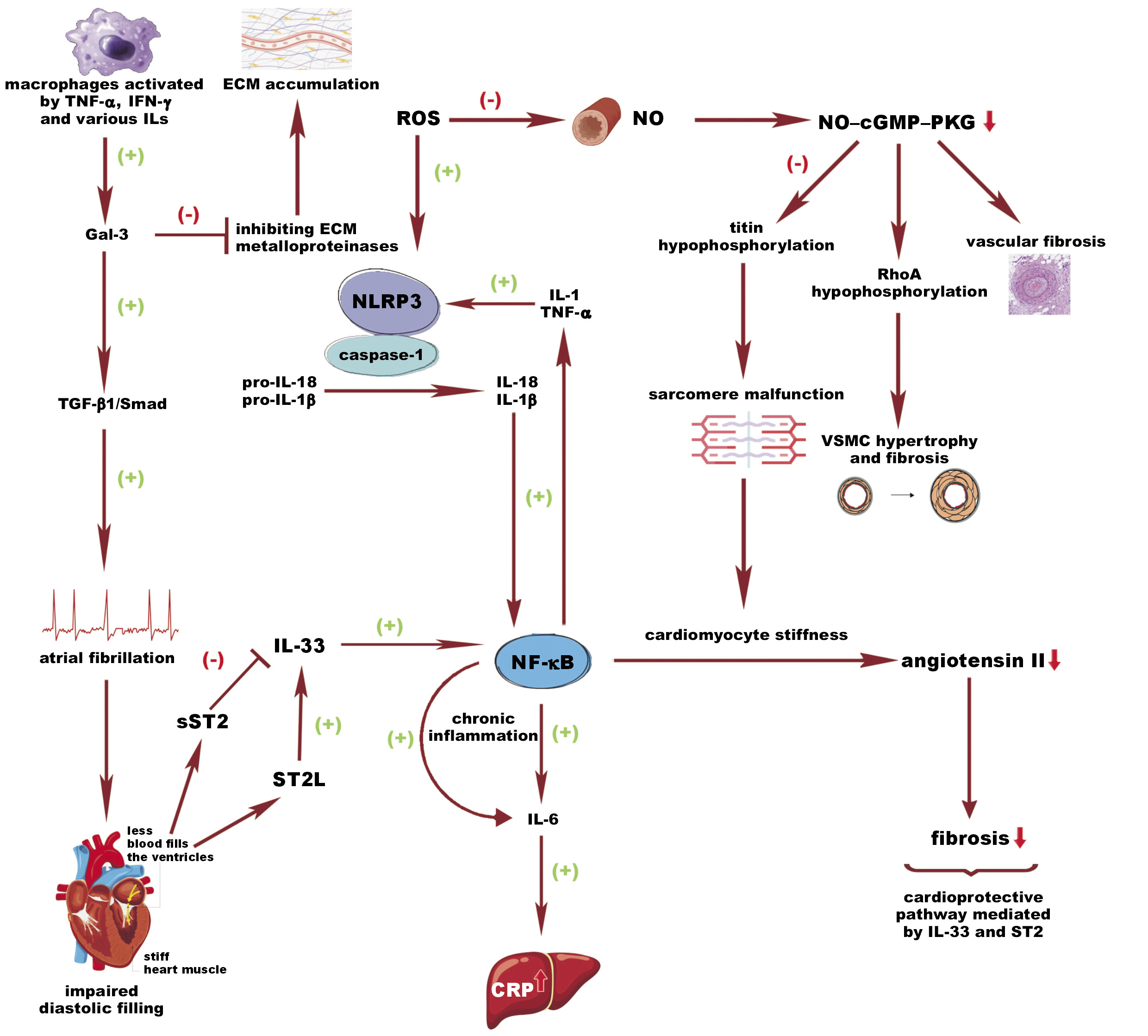
A prominent contributor to this comorbidity-driven inflammation is the nucleotide oligomerization domain-like receptor family pyrin domain containing 3 (NLRP3) inflammasome.36, 37 This cascade is triggered by interleukin (IL)-1 and tumor necrosis factor alpha (TNF-α), or by reactive oxygen species (ROS) with mitochondrial damage, both leading to the increased activation of NLRP3. Consequently, NLRP3, via adaptor protein, binds to the caspase-1, which cleaves pro-IL-1β and pro-IL-18 to their active forms.38 An effect of IL-1β is seen as a further enhancement of nuclear factor kappa B (NF-κB) production and an increase in IL-6 level, directly stimulating the liver to synthesize highly sensitive C-reactive protein (hs-CRP), which is a widely available biomarker of inflammation.39 On the other hand, higher oxidative stress observed in HFpEF contributes to the enhancement in ROS production with concomitant endothelial dysfunction, causing a drop in nitric oxide (NO) synthesis, which is a direct inhibitor of NLRP3. Nevertheless, NO is not only involved in the cross-talk between these pathways; its impaired production results in the suppression of the NO–soluble guanylate cyclase (cGMP)–protein kinase G (PKG) cascade, leading to vasoconstriction and fibrosis.40 Moreover, NO–cGMP–PKG knockdown results in the hypophosphorylation of titin, which is a key sarcomeric protein involved in diastolic cardiomyocyte relaxation, consequently inducing cardiomyocyte stiffness.41 In addition, PKG downregulation results in the hypophosphorylation of the protein RhoA, which reduces its protective role and leads to the hypertrophic and fibrotic activation of vascular smooth muscle cells (VSMCs).41
Another factor that exerts an antifibrotic and antihypertrophic effect on the heart is IL-33, which is released in response to cardiac mechanical stress and injury.42, 43 Importantly, the cardioprotective signaling pathway activated by IL-33 involves suppression of tumorigenicity 2 (ST2) with its 2 variants – transmembrane (suppression of tumorgenicity 2 ligand (ST2L)) located at myocytes, fibroblasts and inflammatory cells, which is activated by IL-33, inducing antihypertrophic and antifibrotic mechanisms that promote adaptive remodeling, and a soluble form (soluble suppression of tumorigenicity 2 (sST2)), which plays the role of the decoy receptor, sequestering IL-33.43 On the other hand, in conditions of cardiac stress or damage, an increased level of sST2 is released, preventing the formation of the ST2–IL-33 complex and suppressing its cardioprotective effect.43 Of note, IL-33 activates NF-κB, which has a dual role in acute hypoxia and cardiac injury and exerts a protective role by inhibiting NF-κB activation induced by angiotensin II, a hypertrophic stimulus.44 However, its chronic activation promotes HF by enhancing the effects of IL-1, TNF-α and IL-6.44
Galectin-3 (Gal-3) is another molecule with a dual role. It is secreted by activated macrophages and plays apoptotic and antinecrotic roles. However, its long-term overexpression observed in HF enhances pro-inflammatory and pro-fibrotic processes.45 Galectin-3, via fibroblast activation, stimulates extracellular matrix (ECM) components like collagen I and collagen III and the synthesis of cytoskeletal proteins, simultaneously inhibiting matrix metalloproteinase (MMP)-induced degradation.45 It is worth mentioning that the disturbance between MMPs and their endogenous inhibitor may lead to excessive accumulation of collagen in the myocardium, which, in turn, further promotes fibrosis and secondary stiffness, ultimately impairing the diastolic function of the ventricles, especially in HFpEF.46 Other factors contributing to collagen degradation are angiotensin II, aldosterone, TNF-α, and transforming growth factor beta (TGF-β).47 Moreover, Gal-3 induces aortic valve calcifications through NF-κB and, by the TGF-β1/Smad pathway, promotes atrial fibrillation (AF), subsequently leading to impaired diastolic filling.48
The cumulative effect of these molecular alterations — inflammation, fibrosis and endothelial dysfunction — leads to structural and functional impairment, characterized by myocardial stiffening and impaired ventricular relaxation, accompanied by increased filling pressures and atrial remodeling. As a result, the clinical manifestation of HF presents as dyspnea, exercise intolerance and systemic congestion. A complex metabolic interplay is demonstrated in Figure 1, and key findings regarding biomarkers and pathways related to SI are summarized in Table 1.
Clinical role of inflammatory biomarkers in HFpEF
Heart failure is a progressive disease that develops through stages, as defined by the American College of Cardiology (ACC) and the American Heart Association (AHA).49
The presence of risk factors contributing to HF development, e.g., T2DM, corresponds to stage A, followed by asymptomatic structural heart diseases (stage B), the presence of clinical symptoms of HF (stage C), and end-stage or refractory HF described as stage D.50 Even though the risk factors of HFpEF are well-defined, the determinants contributing to the progression of HFpEF from stage A to B, as well as more advanced stages, are under investigation.51 Several biomarkers involved in the inflammatory processes underlying HFpEF development are considered to be predictors of disease progression and patients’ outcomes, as well as therapeutic targets.52
A recent meta-analysis emphasized that high levels of CRP were associated with a 9% increase in the risk of HFpEF development.6 In addition, a cohort study revealed the significant role of other NLRP3 inflammasome molecules, particularly IL-6 in HFpEF and TNF-α in the entire HF group.53 Importantly, a prospective study within a PREVEND cohort demonstrated strong evidence of IL-6’s predictive role in new-onset HFpEF, underscoring its significance in HFpEF, but not in HFrEF incidence prognosis.54
However, among patients diagnosed with HFpEF, significantly higher CRP levels were noted in those with a greater comorbidity burden, whereas CRP was within a normal range in 40% of individuals. These findings underscore the need to broaden the spectrum of biomarkers involved in the assessment of patients with HFpEF.55 Even though current HF phenotyping relies primarily on the echocardiographic examination, a meta-analysis involving proteomic studies that compared HFpEF and HFrEF biomarker profiles suggested that they can be distinguished based on higher levels of IL-6 and lower levels of syndecan-1 (SDC-1) and NO in HFpEF patients, highlighting the difference in prevailing pathomechanisms underlying HF presentation.56
Moreover, a meta-analysis on the role of CRP in HF progression confirmed its predictive value regarding cardiovascular and all-cause mortality. Of note, the significance of CRP regarding long-term adverse cardiovascular outcomes was inconclusive, depending on its presentation in the included studies. A significant association was found for categorical variables, while no such effect was observed for continuous variables.6 Nevertheless, the LUCRIC study has analyzed the difference in the inflammatory biomarkers comparing patients with HFpEF and HFrEF, and exhibited the role of IL-6 in the mortality prognosis in a mean 9.9-year follow-up period among HFpEF patients.5 Similar results were observed for hs-CRP, whereas they failed to be significant after full adjustment.5 Of note, neither IL-6 nor hs-CRP predicted cardiovascular mortality in HFrEF patients, hence highlighting the role of subclinical inflammation in the HFpEF population.5 However, recent results from the TOPCAT trial revealed that hs-CRP ≥ 2 mg/L is an appropriate tool to identify HFpEF patients at a higher risk of HF events and cardiovascular mortality.57
Other relevant biomarkers, whose role in the progression to clinically overt HF among patients with risk factors was evaluated in the retrospective analysis of the STOP-HF longitudinal study, are sST2 and Gal-3.58 This study confirmed the role of Gal-3 in the predictive profile, along with B-type natriuretic peptide (BNP) and high-sensitivity troponin I, whereas it rejected sST2 and IL-6. The results in terms of IL-6 were contrary to those of larger, prospective studies.58 The role of Gal-3 was further confirmed by a meta-analysis, which considered it an appropriate biomarker to indicate new-onset HFpEF, as well as a valuable predictor of adverse outcomes and severity of left ventricular diastolic dysfunction (LVDD) in HFpEF.59 Furthermore, Gal-3 is involved in the activation of MMP.46 Among patients with hypertension, MMP-9 and tissue inhibitor of metalloproteinase 1 (TIMP-1) indicated significantly greater degrees of asymptomatic LVDD.46 Galectin-3 also induces the upregulation of TGF-β, whereas its role as a biomarker is modest.46 In contrast to another TGF-β family molecule, substantially increased inflammation and oxidative stress, known as growth differentiation factor 15 (GDF-15), was shown to be an independent risk factor of adverse outcomes across the entire HF spectrum, with its incremental prognostic role in the HFpEF subgroup.60 In another study, the GDF-15 level measured within 48 h of hospital admission in patients with HFpEF was a better predictor of 1-year rehospitalization than N-terminal pro-B-type natriuretic peptide (NT-proBNP).61
Nevertheless, while focusing on sST2 among individuals hospitalized due to HF, its baseline levels are associated with further rehospitalizations and all-cause mortality, independently of ejection fraction. Furthermore, sST2 combined with NT-proBNP, especially in HFpEF patients, is suggested to improve the predictive value of the test.62 Another clinical trial analyzing the role of sST2 noted its role as a significant biomarker in both HF phenotypes, whereas in HFpEF, sST2 revealed a stronger association with patients’ outcomes.63 Other molecules involved in the pathogenesis of HFpEF are mechanistically crucial and play key roles as therapeutic targets, whereas their clinical predictive role is limited and currently of lower importance than the aforementioned parameters.
It is also important to note that SI and the activation of various inflammatory biomarkers or processes can contribute to congestion development, and vice versa.64, 65, 66, 67 Moreover, several mechanisms of both congestion and inflammation contribute to fibrosis and impaired sodium handling, which are part of the disease’s pathophysiology. However, clinical trial data indicates that these pathways are independent. For example, interventions targeting the natriuretic peptide axis do not reduce inflammatory processes.68 Importantly, the role of the lymphatic system has recently been demonstrated in both HF and decongestion, further linking the immune system to congestion development.69, 70, 71, 72, 73, 74
Iron deficiency in patients with HF is also an important biomarker for predicting outcomes.75, 76, 77 Patients with HF are more likely to develop an iron shortage, which can lead to poorer therapeutic results than in patients without such conditions. Jankowska et al. defined iron deficiency as a high (≥1.59 mg/L) level of serum soluble transferrin receptor (sTfR), indicating unmet cellular iron needs, and a low (<14.5 ng/mL) hepcidin level, reflecting low iron storage.75 Patients with acute HF (AHF) who met both criteria had the highest mortality rate of 41%. In comparison, a mortality rate of 15% was noted in individuals with an isolated high sTfR level, and a rate of 7% was observed in those with an isolated low hepcidin level.75 Another trial has connected lactate levels with sTfR. Biegus et al. assessed the relationship between elevated markers (>2 mmol/L and >1.59 mg/L, respectively) in patients with AHF and the occurrence of adverse outcomes.76 It showed that with both markers present, the prognoses were poorer than in patients with one or none of the criteria met.76 The solution to this issue may be iron supplementation. Ahmed et al. conducted a meta-analysis on intravenous (IV) iron supplementation in patients with HF.77 The results indicated that such therapy can reduce CV mortality, 1-year all-cause mortality, first HF hospitalization, and even improve left ventricle ejection fraction (LVEF).77 These studies underscore the importance of assessing iron status in patients with HF, especially those with AHF, as it may be considered a valuable predictive factor of future outcomes. Another reason for its importance is the possibility of reversing it by IV iron supplementation. Although the relationship between iron metabolism and inflammation is well-documented, with evidence pointing to the modulatory role of hepcidin in regulating iron availability, current state of knowledge does not allow us to understand the impact of inflammation on the effectiveness of IV iron therapy. Therefore, additional long-term studies are needed in this area.78
Comorbidities associated with HFpEF and inflammation
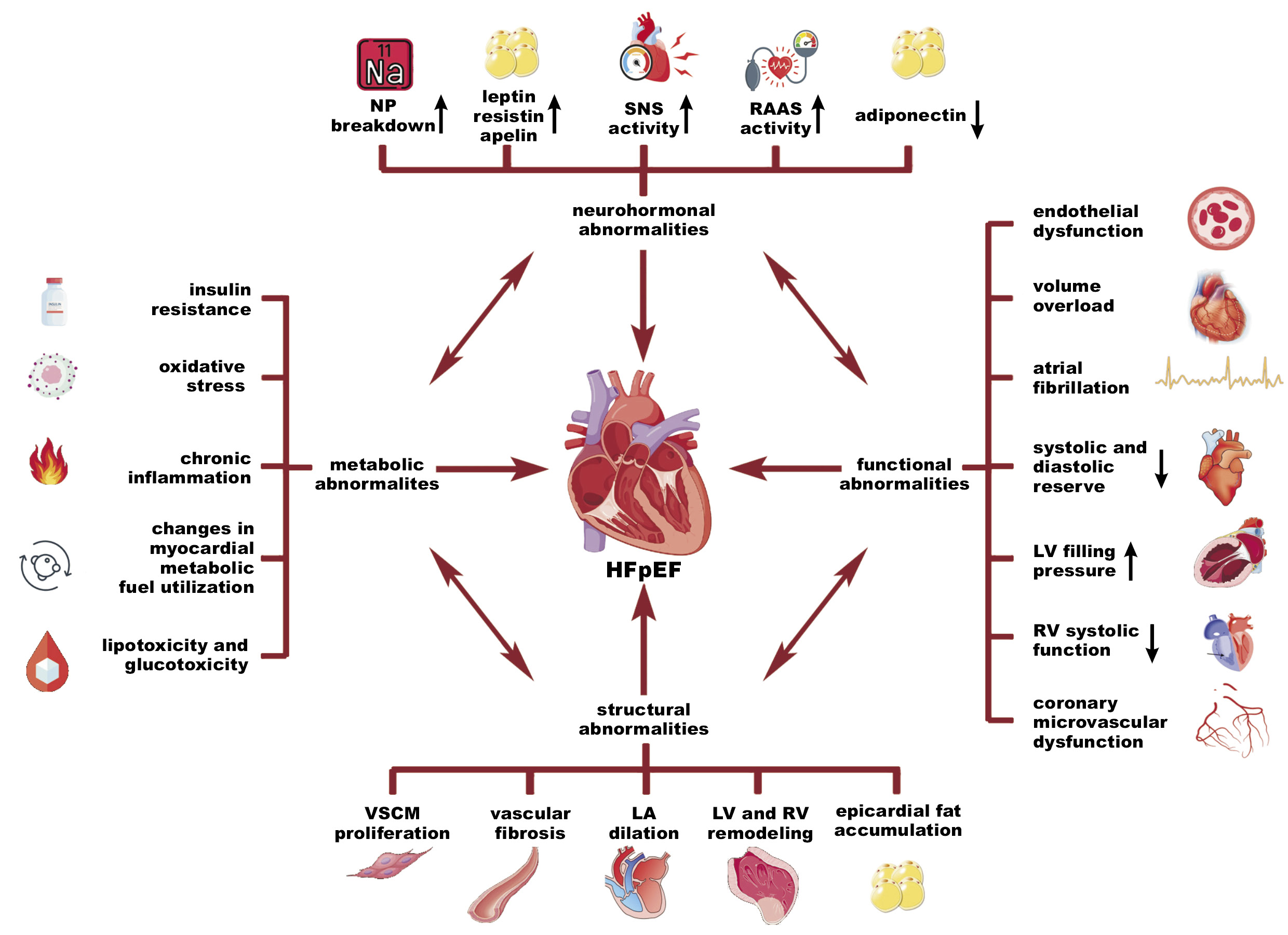
During the past decades, medical attention has concentrated on the management of HFrEF, which resulted in clinical success and a decline in mortality rates within this particular phenotype. The described progress can be attributed to the implementation of a well-established quadruple therapy, consisting of sodium–glucose cotransporter-2 (SGLT-2) inhibitors, mineralocorticoid receptor antagonists (MRAs), beta-blockers, and angiotensin converting enzyme (ACE) inhibitors.2, 79, 80, 81 Despite the improvement in HFrEF survivability, another issue has emerged. Currently, the predominant form of HF is HFpEF. This HF subtype is a surging threat, accounting for more than half of all HF hospitalizations.2, 82 It is a complex condition that creates various diagnostic and therapeutic difficulties due to its multifactorial and heterogeneous etiology, which is still not well understood.2, 14 Thereupon, specific attention should be given to established risk factors that contribute to the development of HFpEF, with a strong focus on the widespread comorbidities associated with this condition.2, 5, 13, 14, 15, 16, 20, 21, 22, 24, 79, 82, 83, 84, 85 Distinguishing each coexisting disorder and a thorough evaluation are essential to assess whether the condition is a complication of HF or an independent disease that can be addressed through an individualized therapeutic strategy.13 Accordingly, it is imperative to investigate the correlation between the onset of HFpEF and the prevalence of comorbidities. We hereby propose a focused examination of low-grade chronic inflammation, which not only contributes to HFpEF evolution but also worsens its course by the promotion of comorbidities (Figure 2).5, 13, 24, 82, 86
Atrial fibrillation
Atrial fibrillation is a common form of arrhythmia, widely diagnosed in patients with HFpEF due to the presence of shared pathophysiological mechanisms. It has been associated with left atrial (LA) wall remodeling and initiated by increased LA pressure, as a consequence of impaired diastolic function of the left ventricle.13, 86, 87 The etiological co-occurrence between these 2 conditions is associated with the activation of maladaptive processes, such as chronic inflammation, oxidative stress and endothelial dysfunction. These risk factors are induced by highly prevalent diseases, mostly hypertension, obesity, T2DM, and chronic kidney disease (CKD). The conditions are manageable in many cases, therefore, they represent an important aspect of curtailing HFpEF progression as well as implementing suitable treatment.87, 88 On a molecular
level, inflammation plays a pivotal role in disease evolution since pro-inflammatory cytokines are responsible for activating fibroblasts that promote remodeling via collagen deposition within an ECM inside the LA myocardium. In patients suffering from HFpEF, AF might occur after HF diagnosis, concurrently, or even prior to HF identification, especially if HFpEF is subclinical or has been stable.86 Regardless of the coincidence of these 2 conditions, AF has been identified as a significant risk factor for mortality when compared to HFpEF patients with sinus rhythm.86 Therefore, it is important to perceive AF not only as a coexisting disease, but also as a substantial stratification factor that may influence therapeutic interventions, including the mitigation of risk factors, proper pharmacotherapy, and the performance of electrophysiological procedures.2, 86
Obesity
Obesity is a condition characterized by excessive fat accumulation.15 The global prevalence of this disease has increased drastically over the past few decades, reaching pandemic levels.15, 89 In 2024, it was estimated that 13% of the adult world population suffered from obesity, while 39% were overweight.15 Accordingly, HF and obesity are two conditions that increasingly coexist each year, and impact each other via various pathophysiological interactions, including structural, functional, metabolic, and neurohormonal abnormalities.13, 14, 15, 16, 79, 82, 90, 91 Evidence suggests a strong interconnection between all phenotypes of HF and obesity; however, the association is remarkable and most significant in terms of HFpEF, where increased body mass is believed to be not only a comorbidity but, most importantly, a direct cause of HFpEF pathogenesis.2, 16, 90
It has been established that adipose tissue is an abundant source of biologically active molecules. Fat accumulation is associated with the dysregulation of various compounds, including leptin, resistin, adiponectin, and apelin.92 These molecules have multidirectional effects, such as appetite regulation, vasodilatation, insulin sensitization, the suppression of inflammation, and the production of ROS.92 The impaired function of these adipokines can contribute to the development of metabolic disorders, such as poor glucose tolerance, increased synthesis of fatty acids with pro-inflammatory potential, elevated production of reactive oxygen and nitrogen species, heightened sympathetic nervous system (SNS) activity, and, above all, the promotion of inflammation mediated by macrophages through the secretion of cytokines.15, 92, 93 This particular situation is a starting point at which metabolic and neurohormonal abnormalities contribute to the development of functional and structural alterations that manifest in the course of HFpEF. It is important to acknowledge the chronic nature of this process, which may remain in the subclinical phase for many years. However, the presence of additional contributing factors, such as AF, can precipitate the development of full-blown HFpEF and result in a condition that may be difficult or even impossible to reverse.13, 14, 15, 16, 79, 90, 93 The impact of inflammation on cardiac remodeling in the HFpEF–obesity subset is extremely complex, but some common structural observations include perivascular and interstitial fibrosis, VSMC proliferation, LV and RV remodeling, LA dilation, and epicardial fat accumulation.15 Lastly, these findings contribute to functional impairment that is characterized by volume overload, coronary microvascular dysfunction, AF, a decline in systolic and diastolic reserve, and an increase in LV filling pressure.15 On this account, obesity plays a crucial role in the pathogenesis of HFpEF, as well as in its management and therapy. Given its reversible nature and the potential for early intervention, especially in patients with an extreme risk of adverse cardiovascular events, it should receive proper attention. Pharmacotherapy plays an essential role in the treatment of obesity, primarily due to the ineffectiveness of lifestyle modification in many cases.
Type 2 diabetes mellitus
Type 2 diabetes mellitus is another example of comorbidity associated with inflammation-driven promotion of HFpEF. During periods of uncontrolled hyperglycemia, the spontaneous formation of advanced glycation end products (AGEs) has been observed.79, 93, 94 These compounds can promote an inflammatory response by activating the receptors for advanced glycation end products (RAGEs), commonly expressed on the surface of endothelial cells, smooth muscle cells and macrophages.95 The binding of AGEs to their receptor triggers a signaling cascade that activates the NF-κB pathway.95 This, in turn, leads to the production and release of pro-inflammatory cytokines, such as TNF-α, IL-1β and IL-6,95 which promote inflammation, cause vascular dysfunction and contribute to HF pathogenesis. A vicious cycle ensues: chronic hyperglycemia leads to increased accumulation of AGEs, resulting in a heightened inflammatory response and cardiovascular damage, which causes deterioration of glucose tolerance.95 Chronic subclinical inflammation results in oxidative stress, endothelial injury, NO signaling deficiency, and altered calcium handling.94, 96 Subsequently, a compensatory response occurs in the form of neurohormonal activation, myocyte hypertrophy and fibrosis. These processes lead to ventricular remodeling, thereby contributing to the onset of HFpEF.79, 87, 94, 96 On account of sympathetic hyperactivity, symptoms such as tachycardia and/or arrhythmias may manifest.96 Therefore, it is important to evaluate the role of T2DM-driven inflammatory changes in the progression of HF, given its reversible character and the potential to counteract the development of HFpEF.2, 14, 87, 97 The significance of SGLT-2 cotransporter inhibitors must be emphasized. The STRIDE trial substantiated the efficacy of semaglutide, demonstrating a 50% reduction in all-cause mortality.27
Treatment
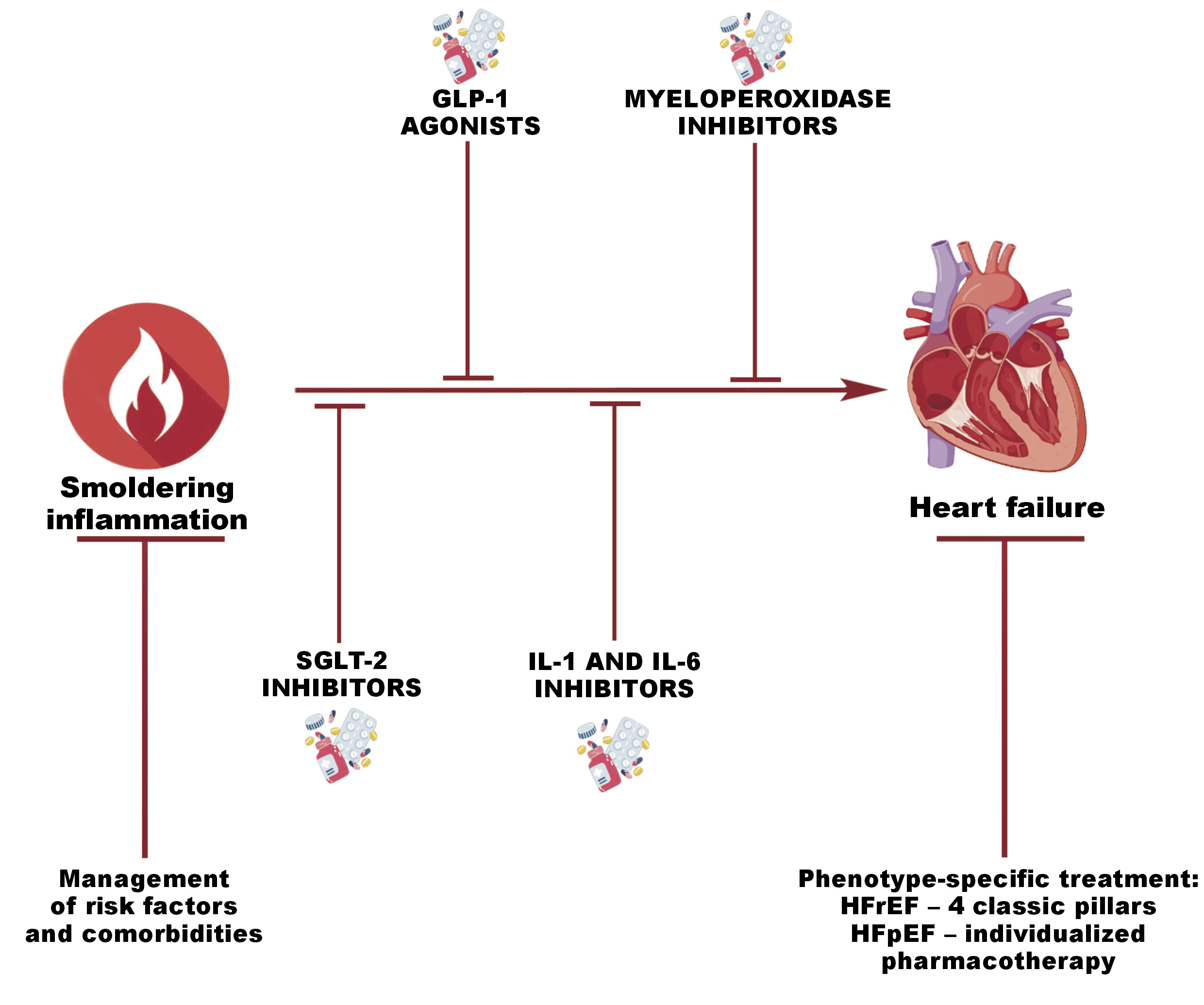
Guideline-directed medical therapy (GDMT) has revolutionized the management of HFrEF, with high-level evidence supporting the use of ACE inhibitors and angiotensin receptor/neprilysin inhibitors (ARNIs), beta-blockers, MRAs, and, more recently, SGLT-2 inhibitors to reduce mortality.98 Emerging studies suggest a potential therapeutic benefit of medication usage in the HFmrEF population.98 In contrast, the treatment options for HFpEF remain limited, with SGLT-2 inhibitors being the only drugs that have demonstrated a clinical effect. Thus, there is a great unmet need for novel therapies that target the pathophysiological mechanisms of HFpEF. The new data concerning systemic inflammation and its potential origins in other systemic diseases may facilitate the identification of the phenotype of HF in patients and the provision of the most effective treatment.
Sodium–glucose cotransporter-2 inhibitors have emerged as promising therapeutic agents for the treatment of HF.33 Their unique anti-inflammatory properties are crucial in the inflammation-based pathophysiology of HFpEF. Moreover, they lower the mortality rate in patients with T2DM.99 In a trial on endothelial cells, it was demonstrated that SGLT-2 inhibitors reduce the expression of NF-κB and MMP-9, thereby lowering the inflammatory pathways.100 Simultaneously, they increase the expression of SIRT6, which plays a role in DNA repair. The utilization of these pharmaceutical agents has been associated with a reduced likelihood of major adverse cardiac events.100 Other clinical trials have noted that SGLT-2 inhibitors lower the hs-CRP levels by over 54% after 1 year of usage in patients with T2DM.101 In light of the EMPEROR-Preserved study findings, special attention should be placed on the implementation of empagliflozin in all HFpEF patients, regardless of the presence or absence of T2DM, given its established efficacy in mitigating HFpEF symptoms and improving prognoses.25
Several studies have demonstrated the efficacy of glucagon-like peptide 1 (GLP-1) receptor agonists, particularly semaglutide, in obese HFpEF patients. The administration of these agents resulted in weight loss, enhanced quality of life, and, most importantly, reduced mortality.2, 16, 27, 79, 102, 103 This establishes an important direction in the comprehensive treatment of patients with HF, emphasizing an approach in which obesity is treated as an independent co-occurring disease requiring treatment rather than placing the entire responsibility for alleviating obesity on the patient, which frequently proves unfeasible.
Another area of investigation aimed at finding a more effective method to phenotype patients is myeloperoxidase (MPO) inhibition. During the SATELLITE trial, AZD4831, the MPO inhibitor, has been administered to patients.104 Afterward, the biomarker pathways most related to clinical outcomes in individuals with HFpEF were found to be downregulated. Nevertheless, the cohorts included in this trial were small; therefore, the results must be investigated in a larger group.104
The NLRP3 inflammasome is the next target of anti-inflammatory therapy. Many molecules, including colchicine, GDC-2394 and dapansutrile, are in the stage of clinical development.26, 104, 105, 106 Despite this, colchicine exhibits a strong anti-inflammatory action by targeting the NLRP3 inflammasome and inhibiting the production of pro-inflammatory cytokines, such as IL-1 and IL-6.88, 105 In HFpEF, this property has the potential to reduce SI and possibly regulate maladaptive myocardial remodeling, along with enhancing cardiac function.88 Nevertheless, pro-inflammatory cytokines (IL-1β or IL-6), which are associated with the NLRP3 pathway, have undergone more advanced trials concerning their inhibitors. Anakinra and canakinumab, the inhibitors of IL-1β, have been found to reduce inflammation in patients with CVD. Anakinra has significantly decreased CRP levels in patients with HFpEF.107 The IL-6 inhibitors, namely tocilizumab (ASSAIL-MI) and ziltivekimab (RESCUE) have reduced systemic inflammation, as evidenced by decreased CRP levels in patients with myocardial infarction and CKD, respectively.108, 109 Lastly, the recent CORTAHF trial has shown that the use of burst steroid therapy resulted in lower inflammation, more effective decongestion, and improvements in quality of life in the AHF population.68, 110, 111, 112, 113 Specifically, this study has demonstrated that 7-day therapy with prednisone at a dose of 40 mg in patients with AHF and elevated hs-CRP led to a reduction in hs-CRP levels on day 7 of therapy, and additionally, on day 90, patients who received burst therapy showed a significantly lower risk of rehospitalization or HF decompensation.68, 110, 111, 112, 113 The graphical summary of therapeutic modalities is demonstrated in Figure 3 and summarized in Table 2.
Smoldering inflammation and oral connections in cardiovascular disease
Recent findings support a correlation between SI and oral diseases.114, 115, 116, 117, 118, 119, 120, 121, 122 Numerous studies have demonstrated that chronic, low-grade inflammation accompanies periodontal disease and is associated with oral microbiome imbalances. These imbalances can contribute to a systemic inflammatory burden, directly increasing cardiovascular risk.114, 115, 116, 117, 118, 119, 120, 121, 122 Smoldering inflammation may be a potential mechanistic link between oral health and CVDs, including HF.119, 120 Periodontal diseases, especially periodontitis, contribute to endothelial dysfunction, oxidative stress and vascular remodeling by mediating inflammation, thereby promoting CVD progression.116 Additionally, oral microbiome dysbiosis, characterized by the entry of pathogenic bacteria and their metabolic products into the circulation, can trigger systemic immune pathway responses and induce SI of distant tissues, which amplifies the negative health outcomes associated with SI.122 The translocation of pathological bacteria may occur during processes such as toothbrushing, flossing and chewing, resulting in minor mechanical injuries in the oral cavity, which are the gateway for bacterial dissemination throughout the vascular system.115 Specific oral bacterial pathogens, such as Porphyromonas gingivalis, have been directly implicated in the initiation and progression of atherosclerotic lesions, further substantiating a definitive microbial contribution to cardiovascular risk.115 What is more, overlapping risk factors, such as smoking, obesity and unhealthy diets, sustain persistent inflammatory pathways by establishing optimal conditions for the development of pathological microbiota that promote shared risk between the oral cavity and cardiovascular systems.114 Clinical studies demonstrate that the treatment of oral health conditions (e.g., periodontal disease) may lead to a decreased systemic inflammatory burden and a reduced incidence or progression of CVD.117, 120, 121 Therefore, the maintenance of proper oral health should be prioritized, especially among patients at risk of developing CVD.
Limitations
This review is subject to several limitations. The narrative style of the review, rather than a systematic or meta-analysis approach, raises the possibility of selection bias for the evaluated studies. The second limitation is the heterogeneity of HFpEF phenotypes and variations in study design, study population and biomarker assessment methodology that limit interpretations and generalizability of the findings. The third limitation pertains to the fact that, despite our efforts to provide a comprehensive overview of novel anti-inflammatory therapies, many of the interventions encompassed by this review are based on early-phase trials or relatively small cohorts. Therefore, the long-term efficacy and safety profiles of several interventions remain unknown. The final limitation regards the evolving nature of the field, which may have resulted in the omission of studies that were published at the time of the literature search. In summary, despite these limitations, this review provides context for SI in HFpEF and has identified important considerations for subsequent research.
Clinical implications and future research
Smoldering inflammation plays a crucial role in the pathogenesis of HFpEF, and it should not be overlooked during the diagnosis. The findings summarized in this article can convince clinicians to adopt a more holistic approach to patient care, rather than a narrow focus on one particular disease. Treating the underlying causes of inflammation can positively influence the course of HFpEF. The assessment of the levels of inflammatory markers is a crucial step in the diagnostic process for patients with various medical conditions. Subsequent research on inflammation in HFpEF may result in an update of the guidelines concerning its treatment.
Conclusions
Despite the presence of many known risk factors, recent research has examined the correlation between low-grade chronic inflammation, known as SI, and HF, from its onset to the end stage. A correlation between proinflammatory cytokines, such as IL-1β, IL-6, TNF-α, the NF-κB inflammatory pathway, and serum level of hs-CRP, and its impact on the remodeling of heart tissue has been proven. These factors, along with ROS and other proinflammatory molecules and pathways, lead to hypertrophy and fibrosis, therefore impairing heart muscle function and resulting in HF. Other comorbidities related to inflammation, such as AF, T2DM and obesity, have also been associated with the onset of HF, mainly HFpEF. Therefore, treating these conditions with anti-inflammatory medications may result in the improvement during the course of HF. Moreover, trials on the efficacy of other inflammatory pathway inhibitors have provided promising outcomes in small clinical groups, suggesting the potential of these agents in HF treatment. The described pathophysiological mechanisms of SI demonstrate the complexity of the subject but also emphasize the importance of a thorough understanding of the topic in order to implement proper treatment. Smoldering inflammation begins as subclinical myocarditis and can remain unnoticed for a long time, finally progressing to post-inflammatory dilated cardiomyopathy. Therefore, anti-inflammatory interventions, when administered in conjunction with standard HF treatment, may contribute to the modulation of this progression. Further studies are necessary to optimize timing, dosage and patient selection for maximal benefit.
Ethics approval and consent to participate
Not applicable.
Data availability
Not applicable.
Consent for publication
Not applicable.
Use of AI and AI-assisted technologies
AI-assisted technology (Grammarly; Grammarly Inc., San Francisco, USA) was used for language editing and text refinement. The authors have reviewed and approved all content.